Design and Synthesis of Disk-liked Benzotrithiophene Small Molecules for the Organic Solar Cells Material
LI-Chuan WU 1, Xin-Yi Chen 2 and Chih-Hung Lin 3*
and Chih-Hung Lin 3*
1Department of Applied Chemistry and Material Sciences, Fooyin University, 151 Jinxue Road., Daliao, Kaohsiung City, Taiwan, Republic of China.
2Department of Cosmetic Science, Chang Gung University of Science and Technology, 261 Wen-Hwa 1st Road, Kwei-Shan, Tao-Yuan, Taiwan, Republic of China.
3Center for General Education, Chang Gung University of Science and Technology, 261 Wen-Hwa 1st Road, Kwei-Shan, Tao-Yuan, Taiwan, Republic of China.
Corresponding Author E-mail: chlin@mail.cgust.edu.tw
DOI : http://dx.doi.org/10.13005/ojc/400501
Article Received on : 04 Sep 2024
Article Accepted on : 04 Oct 2024
Article Published : 05 Nov 2024
Reviewed by: Dr. Shireen R Mohammed
Second Review by: Dr. Wayan Sutapa
Final Approval by: Dr. Abdelwahab Omri
This study explores the design and synthesis of small molecules specifically tailored for organic solar cells, aiming to enhance their solubility, film-forming properties, and thermal stability through strategic material design. By achieving these improvements, we sought to advance the technology of organic solar cells. We successfully designed and synthesized a triangular disc-shaped small molecule material with benzotrithiophene as the core. To accurately determine the structure and properties of the target compound, we employed advanced characterization techniques, including TGA, NMR, and DSC. The results confirmed that the material exhibited high purity, thermal stability, and solubility. These remarkable attributes suggest significant potential for the material's use as an active layer in solar cells, paving the way developing of high-performance organic solar cells and opening new avenues in the field.
KEYWORDS:Benzotrithiophene; Organic Solar Cells; Small Molecules; Stille Coupling Reaction; Thermal Stability
Download this article as:| Copy the following to cite this article: WU L. C, Chen X. Y, Lin C. H. Design and Synthesis of Disk-liked Benzotrithiophene Small Molecules for the Organic Solar Cells Material. Orient J Chem 2024;40(5). |
| Copy the following to cite this URL: WU L. C, Chen X. Y, Lin C. H. Design and Synthesis of Disk-liked Benzotrithiophene Small Molecules for the Organic Solar Cells Material. Orient J Chem 2024;40(5). Available from: https://bit.ly/4eith1S |
Introduction
As technology rapidly advances, the energy demand continues to grow. Fossil fuels, including natural gas, oil, and coal, are the most consumed energy sources worldwide. However, their limited reserves and the environmental damage caused by carbon dioxide emissions, such as the greenhouse effect, highlight the need for alternative energy solutions. Renewable energy sources like solar, wind, hydropower, and biomass have become increasingly important. Among these, solar energy is a key area of research.
Solar cells can be classified into three types based on materials: silicon-based, semiconductor, and organic solar cells. Silicon-based cells have a conversion efficiency of 8% to 25%, but they are expensive due to the high cost of silicon and the complex manufacturing process. Semiconductor cells, which include materials like cadmium telluride and copper indium gallium selenide, range from 10% to 32% efficiency and are even more costly. Organic solar cells, though less efficient at 8% to 12%, are the least expensive due to their simpler production processes and use of less expensive materials1.
Due to the high costs of silicon and semiconductor cells, research on organic solar cells has gained momentum. Organic cells are divided into dye-sensitized and organic thin-film cells, with advantages such as low cost, simple production, scalability, flexibility, and transparency, making them a promising future technology2-6. Organic thin-film cells, in particular, offer the potential for lightweight and flexible solar panels that can be integrated into a variety of surfaces, including windows and clothing.
In 1986, Dr. Ching W. Tang developed the first organic solar cell with 1% efficiency7, followed by a bulk heterojunction design by Heeger’s team in 1995, which improved efficiency to 2.9%8. Since then, research has advanced significantly. Organic thin-film solar cells can be classified into polymer-based and small molecule-based types. Polymer-based cells, which use conjugated polymers as the active layer, have seen substantial efficiency improvements but often face issues with stability and scalability. On the other hand, small molecule materials, while typically less efficient, are easier to purify and produce with higher stability and reproducibility.
Recent advancements have led to small molecule materials achieving up to 8% efficiency[10], marking a significant improvement over earlier technologies. These materials, such as copper phthalocyanine and perylene diimides, are being optimized to enhance performance and durability. Innovations in material science, such as the development of new electron acceptors and the engineering of better interfaces, are driving the efficiency of organic solar cells higher. Additionally, research into tandem cell structures, where organic cells are combined with other types of solar cells, has the potential to push efficiencies even further.
The outlook for organic solar cells is optimistic, with ongoing research pushing the boundaries of materials, fabrication methods, and device designs. Efforts are focused on enhancing the performance of organic solar cells to rival traditional photovoltaic technologies, positioning them as a flexible, cost-effective, and eco-friendly substitute for fossil fuels.
Material and Methods
The identification and characterization of all the products were carried out using the following instruments.
Nuclear Magnetic Resonance Spectroscopy (NMR): The Agilent Unity-400 NMR spectrometers were used. D-chloroform served as the solvent for the experiment. For proton spectra, δ = 7.26 ppm (d-CHCl3) was utilized as the internal reference, while for the carbon NMR spectra, δ = 77.16 ppm (CDCl₃) was used as the reference point.
Differential Scanning Calorimeter (DSC): The low-temperature environment was provided using the TA Instruments Unpacking the Q series DSC and RCS cooling system. Samples weighing 3 to 5 mg were first cooled to 0°C and allowed to equilibrate, then heated to a temperature below the sample’s thermal degradation temperature (80°C), and this process was repeated twice.
Thermal Gravimetric Analyzer (TGA): The Perkin Elmer Pyris Thermogravimetric Analyzer was used. Samples, each weighing between 2 to 5 mg, were subjected to an initial thermal treatment to ensure the removal of any volatile components. The samples were subjected to heating to 110°C for 10 minutes while a nitrogen flow of 100 mL/min was maintained. This step was crucial for eliminating moisture and other low-boiling solvents that might otherwise interfere with the thermal analysis. Following this initial heating phase, the temperature was reduced to 50°C to allow the samples to stabilize in a controlled environment. After stabilization, the samples were heated at a programmed rate of 10°C per minute until they reached 750°C. During this process, the sample weight was continuously monitored, and a TGA curve was generated plotting temperature versus the percentage of remaining weight. This curve is used to assess the thermal stability and decomposition behavior of the sample.
The thermal decomposition temperature (Td) was determined by identifying the temperature at which the sample retained 95% of its initial weight. This value provides insight into the thermal stability and the onset of significant weight loss, which often corresponds to decomposition or structural changes in the sample.
All chemicals used were sourced from various suppliers such as Aldrich and Merck, and were used directly without further purification. All solvents utilized in the experiments were sourced from Merck and Fischer. Anhydrous tetrahydrofuran, anhydrous toluene, and anhydrous dichloromethane were all dehydrated using the SD-300 anhydrous solvent purification system.
Synthesis
2,4,6-trichloroaniline (Compound 1)
In a 450 mL two-necked flask, aniline (7 g, 75.2 mmol) and acetonitrile (140 mL) are added. A reflux apparatus is set up and purged with nitrogen. The mixture is brought to 90°C and maintained under reflux. N-chlorosuccinimide (NCS, 30 g, 225 mmol) is weighed out in three equivalents and added in three portions. First, 1 equivalent is added, and the reaction proceeds for 1 hour. Subsequently, another equivalent is added each hour, making three total additions. After the final addition, the reaction continues for an additional hour, and progress is monitored using TLC.
Once the reaction is finished, let the mixture cool to room temperature. Then, add 200 mL of deionized water and stir the mixture vigorously for 1 hour, during which a significant amount of purple solid precipitates. Collect the solid using vacuum filtration, then extract it with 200 mL of diethyl ether. Wash three times with 200 mL of deionized water. Combine the organic layers and dry with anhydrous MgSO4. After drying, filter the solution and evaporate the organic solvent under reduced pressure. Purify the crude product with silica gel column chromatography, using hexane as the eluent. After impurities are removed, a white solid (compound 1) is obtained (13.9 g, 94% yield).1H NMR: 7.2 (s, 2 H), 4.43 (s, 2 H).
1,3,5-trichlorobenzene (Compound 2)
Compound 1 (2 g, 10.18 mmol) is introduced into a 150 mL single-neck flask and dissolved in 10 mL of 70% sulfuric acid aqueous solution. Dissolve NaNO₂ (1.4 g, 20.36 mmol) in 2.5 mL of deionized water. This solution is slowly introduced dropwise over 30 minutes at 0°C using an ice bath. Orange fumes may be observed during the addition. Once the addition is finished, the mixture is agitated at 0°C for 30 minutes. Next, add 15.5 mL of 30% aqueous hypophosphorous acid dropwise over 20 minutes at 0°C, then let the mixture warm to room temperature and stir for 12 hours. The mixture is poured into 100 mL of deionized water, then filtered under reduced pressure to obtain a light orange solid, identified as compound 2 (1.6 g, 88% yield).1H NMR: 7.27 (s, 3 H).
1,3,5-trichloro-2,4,6-triiodobenzene (Compound 3)
In a 25 mL single-necked flask, H₅IO₆ (534 mg, 2.34 mmol) is dissolved in 10 mL of conc. H2SO4. Add 2.1 g of iodine and stir at room temperature for 30 minutes. Then, keep the temperature at 0°C with an ice bath and slowly add 500 mg of compound 2. After the addition, let the reaction proceed at room temperature for 3 days. Afterward, Pour the mixture over crushed ice and gradually add aqueous sodium hydroxide until the mixture is neutralized and the color changes. The crude solid is collected by filtration under reduced pressure and then washed with methanol three times with deionized water. The product is subsequently recrystallized from tetrahydrofuran, yielding a white needle-like solid, compound 3 (893 mg, 58% yield).MS (EI, C6Cl3I3):calcd, 559.14;found, 559.7.
((2,4,6-trichlorobenzene-1,3,5-triyl)tris(ethyne-2,1-diyl))tris (trimethylsilane) (Compound 4)
Compound 3 (2.21 g, 3.95 mmol) and CuI (135 mg, 0.71 mmol) are placed into a 100 mL two-neck flask. Within a glovebox, the catalyst Pd(PPh3)4 (411 mg, 0.355 mmol) is weighed and introduced into the flask. The reaction setup is equipped for reflux under a nitrogen atmosphere, and tetrahydrofuran (18 mL) along with diisopropylamine (2 mL) are degassed and introduced. After stirring, trimethylsilylacetylene (2.81 mL, 19.75 mmol) is added. The mixture is brought to 70°C and refluxed for 30 hours. After quenching the reaction with deionized water, remove tetrahydrofuran under reduced pressure. Extract the residue with 100 mL of dichloromethane and wash it with 100 mL of deionized water three times. The purification process follows the procedure described for compound 1. The purified compound is isolated as a white solid, compound 4 (984 mg, 53% yield).1H NMR: 0.29 (s, 27 H).
benzotrithiophene (Compound 5)
Compound 4 (1.9 g, 4.04 mmol) and Na₂S·9H₂O (5.82 g, 24.2 mmol) are placed into a 300 mL single-neck flask, to which 100 mL of N-methyl-2-pyrrolidone is added as the solvent. Reflux the mixture at 185°C under nitrogen for 12 hours, then cool to room temperature. It is then poured into 300 mL of saturated ammonium chloride solution, and the solid is filtered by gravity. The product is subsequently purified by silica gel column chromatography with hexane as the eluent. The purified compound is a white crystalline solid, compound 5 (534 mg, 53.6% yield). 1H NMR: 7.64 (d, J = 5.6 Hz, 3 H), 7.54 (d, J = 5.2 Hz, 3 H).
2,5,8-tris(trimethylstannyl)benzotrithiophene (Compound 6)
Compound 5 (531 mg, 2.15 mmol) is added to a 100 mL two-neck flask, which is evacuated under vacuum and flushed with nitrogen three times before adding 45 mL of anhydrous tetrahydrofuran. Slowly add n-BuLi (2.5 M, 5.17 mL, 12.9 mmol) to the ice-cold mixture. Stir at room temperature for 1 hour. Subsequently, trimethyltin chloride (1.0 M, 15.1 mL, 15.1 mmol) is added to the mixture, still maintaining the ice bath. After returning to room temperature, the reaction proceeds for 14 hours. The reaction is quenched with deionized water and then concentrated under reduced pressure to remove organic solvents. Extraction is performed with dichloromethane (100 mL) and deionized water (100 mL × 3). The product is purified as described for compound 1 and recrystallized from methanol to yield a white solid, compound 6 (869 mg, 55% yield).1H NMR: 7.69 (m, 3 H), 0.48 (m, 27 H).
5-bromothiophene-2-carbaldehyde (Compound 7)
In a 100 mL two-neck flask, DMF (10.6 mL) is added under a nitrogen atmosphere and an ice bath. Gradually add POCl₃ (14 mL, 153.3 mmol) and stir at room temperature for 1 hour. Following this, introduce 6 mL (10.04 g, 61.6 mmol) of 2-bromothiophene, then heat the mixture to 80°C and reflux for 24 hours. To quench the reaction, the mixture is poured over a large quantity of crushed ice. Extraction is performed with ethyl acetate (100 mL) and sodium hydroxide solution (100 mL × 3). Purification follows the procedure described for compound 1, yielding a pale yellow liquid, compound 7 (10.5 g, 89% yield), was obtained. 1H NMR: 9.78 (s, 1 H), 7.51 (d, J = 4Hz, 1 H), 7.19 (d, J = 4Hz, 1 H).
2-ethylhexyl (E)-3-(5-bromothiophen-2-yl)-2-cyanoacrylate (Compound 8)
Place compound 7 (500 mg, 2.62 mmol) into a 25 mL two-neck flask. Under a nitrogen and with a reflux setup, add dry CHCl3 (10 mL). Add 2–3 drops of triethylamine and 2-ethylhexyl cyanoacetate (2.76 mL, 13.07 mmol). Allow the reaction to proceed at room temperature for 12 hours. Quench with deionized water, then extract with 100 mL of CH2Cl2 and wash three times with 100 mL of deionized water. Purify the product as described for compound 1. Recrystallize the pure product from methanol to obtain compound 8(808 mg, 83% yield). 1H NMR: 8.20 (s, 1 H), 7.51 (d, J = 4 Hz, 1 H), 7.19 (d, J = 4 Hz, 1 H), 4.22 – 4.20 (m, 4 H), 1.71 – 1.68(m, 1 H), 1.45 – 1.3 (m, 8 H), 0.94 – 0.89 (m, 6 H); 13C NMR: 162.59, 145.62, 137.72, 137.51, 131.45, 124.2, 115.45, 99.59, 69.0, 38.74, 30.27, 28.88, 23.71, 22.91, 14.01, 10.98.
(2E,2’E,2”E)-tris(2-ethylhexyl) 3,3′,3”-(5,5′,5”-(benzo[1,2-b:3,4-b’:5,6-b”]trithiophene-2,5,8-triyl)tris(thiophene-5,2-diyl))tris(2-cyanoacrylate) (Compound 9)
Place compound 6 (150 mg, 0.204 mmol) and compound 8 (453 mg, 1.22 mmol) into a 50 mL two-neck flask. Within a glovebox, weigh and add the catalyst Pd(PPh3)4 (12 mg, 0.01 mmol). Set up the apparatus for reflux, degas the solvent toluene (15 mL), and add it to the flask. Heat to 100°C and reflux for 3 days. Quench the reaction with deionized water and extract with dichloromethane (100 mL) and deionized water (100 mL x 3). Purify the product as described for compound 1, using hexane and dichloromethane (1:1) as the initial eluent for column chromatography. After the first fraction elutes, increase the polarity of the eluent gradually, using hexane and dichloromethane (1:4) for gradient elution. Reprecipitate the purified product from a tetrahydrofuran and methanol mixture to isolate a bright orange solid, compound 9 (90 mg, 40% yield). 1H NMR: 8.06 (s, 3 H), 7.54 (d, J = 4 Hz, 3 H), 7.21 (d, J = 3.6 Hz, 3 H), 6.95 (s, 3 H), 4.23 (m, 6 H), 1.77 (m, 3 H), 1.5 – 1.39 (m, 24 H), 1.02 – 0.96 (m, 18 H); 13C NMR: 162.26, 145.91, 145.34, 140.01, 135.42, 135.04, 130.95, 130.51, 125.36, 118.60, 115.44, 98.02, 69.14, 38.64, 30.38, 28.98, 23.79, 23.04, 14.13, 11.0; MS (FAB+, C60N3S6O6):calcd, 1114.55;found, 1113.9.
Results and Discussions
The design of active layer materials in organic thin-film solar cells directly influences their power conversion efficiency. Recent advancements in research have focused extensively on optimizing the structural design of these materials to boost their performance. When designing organic materials, the following five key aspects need to be considered 11: 1. Intra-chain and inter-chain charge transfer can be enhanced by pairing donor materials with high highest occupied molecular orbital (HOMO) levels with acceptor materials that have low lowest unoccupied molecular orbital (LUMO) levels. This combination promotes effective charge transfer between the donor and acceptor, leading to a reduction in the energy gap. 2. Aromaticity: The planar configuration of aromatic rings in the polymer backbone allows for high delocalization of π-electrons, which can effectively lower the energy gap. 3. Substituent effect: Incorporating electron-donating groups into the donor molecules can increase the HOMO level while adding electron-withdrawing groups to the acceptor molecules can decrease the LUMO level. This approach enhances the interaction between the donor and acceptor, effectively narrowing the energy gap. 4. Intermolecular interactions: Enhancing the double-bond characteristics between each donor and acceptor can result in the creation of more stable quinoid structures. as shown in figure 111, which influences intermolecular interactions and reduces the energy gap. 5.π-Conjugation length: Increasing the length of π-conjugation effectively lowers the material’s energy gap.
 |
Figure 1: Thiadiazole’s resonance structures |
In this study, we developed a small molecule featuring oligothiophenes to function as the active layer material in organic thin-film solar cells. Oligothiophenes are commonly employed in organic semiconductor applications because of their excellent charge mobility. Additionally, the ability to adjust the number of thiophene rings or the placement of side chains during synthesis allows for precise tuning of electronic energy levels12. To achieve high-efficiency organic photovoltaic devices, the oligothiophene molecules need to exhibit a low energy gap, broad absorption spectrum, and suitable energy levels. One effective approach is to introduce electron-withdrawing groups into their conjugated long chains, enhancing π-electron polarizability and conjugation, which can widen the absorption spectrum and enhance the donor-acceptor interaction, thus promoting intramolecular charge transfer.
Therefore, this study proposes a nearly disc-shaped triangular small molecule structure based on benzotrithiophene (BTT) as the core. Compared to polymer structures, small molecule designs offer more diversity. In addition to the usual linear structures, the literature also mentions dendritic, star-shaped, and other configurations of small molecules 13-17, each with different conversion efficiencies and parameter characteristics. Generally, enhancing the planarity of two-dimensional structures is beneficial for molecular stacking, which facilitates π-electron transitions and increases charge mobility. The use of benzotrithiophene as a core structure in small molecules is not commonly seen in current literature. To explore the relationship between its structure and optoelectronic properties, we designed a molecule using benzotrithiophene as the core, with thiophene groups at the three terminals to increase conjugation, and 2-ethylhexyl cyanoacetate groups on the periphery as electron acceptors. The resulting small molecule is (2E,2’E,2”E)-tris(2-ethylhexyl) 3,3′,3”-(5,5′,5”-(benzo[1,2-b:3,4-b’:5,6-b”]trithiophene-2,5,8-triyl)tris(thiophene-5,2-diyl))tris(2-cyanoacrylate) (compound 9). The chemical structure is illustrated in the figure 2. To improve the solubility of the small molecule, we selected 2-ethylhexyl cyanoacetate over 3-ethylrhodanine, utilizing the long carbon chain of 2-ethylhexyl cyanoacetate to enhance solubility, which in turn facilitates synthesis and device fabrication.
 |
Figure 2: Schematic diagram of the molecular structure design of compound 9 |
 |
Scheme 1: Synthesis of compounds |
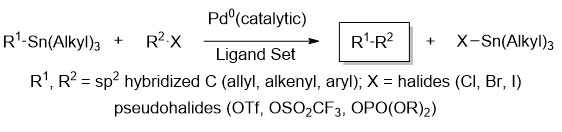
Scheme 1 illustrates the molecular structure and the typical synthetic methods for these compounds. The synthesis of the compound 9 small molecule begins with the preparation of the benzotrithiophene (BTT) core. Initially, aniline is chlorinated at positions 2, 4, and 6 using N-chlorosuccinimide to obtain a chlorinated intermediate. This intermediate is then reacted with sodium nitrite (NaNO₂) and sulfuric acid to form a diazonium salt. Hypophosphorous acid (H₃PO₂) is used to replace the diazonium group with a hydrogen atom, resulting in the precursor compound 2 (1,3,5-trichlorobenzene). Subsequently, compound 2 is iodinated at positions 2, 4, and 6 to yield compound 3. A Sonogashira coupling reaction[18] is employed to attach a trimethylsilyl group to the iodinated positions, followed by cyclization using sodium sulfide nonahydrate to form benzotrithiophene compound 5. Compound 6 is then synthesized by reacting n-butyllithium with compound 5 at the 2-position of the thiophene rings and attaching trimethyltin. The synthesis of compound 8 starts with the reaction of 2-bromothiophene with Vilsmeier reagent to introduce an aldehyde group at the 5-position. This is followed by a Knoevenagel condensation with 2-ethylhexyl cyanoacetate to obtain compound 8. Finally, compound 6 and compound 8 undergo a Stille coupling reaction19 to form the target compound 9. In this experiment, the organic metal-catalyzed reactions used include the Sonogashira coupling reaction and the Stille coupling reaction. Different coupling reactions are selected based on the reactivity of the carbon-carbon single bonds to be synthesized. Below is a simplified schematic of the Stille coupling reaction.
As an active layer material for organic solar cells, high thermal stability is essential. Therefore, this study uses TGA and DSC to measure the thermal properties of the synthesized small molecule materials. Td of Compound 9 was measured to be 381°C, as shown in Figure 3; the melting point is 275°C, and the crystallization temperature is 264°C, as shown in Figure 4. When designing the molecular structure of Compound 9, each end features a single thiophene ring, resulting in a very rigid planar disc-like structure. Consequently, its melting point reaches 275°C. The high melting point helps prevent temperature increases during prolonged use of the device, which could otherwise affect molecular morphology. Additionally, during the cooling process, besides a single exothermic peak representing the crystallization point, no other exothermic peaks were observed. This indicates that there are no other phase transitions, and the compound exhibits high thermal stability, making it well-suited for use as an active layer material in solar cells.
 |
Figure 3: TGA thermograms of compound 9 |
 |
Figure 4: DSC thermograms of compound 9 |
Conclusion
In this study, we successfully created a small molecule with a triangular disc-shaped small molecule structure based on benzotrithiophene (BTT) as the core, with thiophene and 2-ethylhexyl cyanoacetate as electron acceptors at the terminals. The target compound (Compound 9) was successfully synthesized through nine reactions, including coupling reactions. This compound dissolves in organic solvents such as tetrahydrofuran, chloroform, chlorobenzene, and ortho-dichlorobenzene, making it well-suited for spin-coating applications. From the perspective of thermal stability, the compound exhibits a decomposition temperature (Td) as high as 381°C, indicating excellent thermal stability. Differential scanning calorimetry results show that the compound does not exhibit any phase transitions, underscoring its excellent thermal stability and its potential as an active layer material for solar cells. In the future, we plan to perform comprehensive analyses of the compound’s optical and electrochemical properties, aiming to establish its effectiveness as an active layer material in organic solar cells.
Acknowledgment
This research was funded by the Chang Gung University of Science and Technology.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
References
- Green, M. A.; Emery, K.; Hishikawa, Y.; Warta, W.; Dunlop, E. D. Prog. Photovolt: Res. Appl. 2013, 21 (1), 1-11.
CrossRef - Tang, C. W.; Albrecht, A. C. Nature 1975, 254 (5500), 507-509;
CrossRef - Ghosh, A. K.; Morel, D. L.; Feng, T.; Shaw, R. F.; Rowe, C. A. J. Appl. Phys. 1974, 45 (1), 230-236.
CrossRef - Merritt, V. Y.; Hovel, H. J. Appl. Phys. Lett. 1976, 29 (7), 414-415;
CrossRef - Morel, D. L.; Ghosh, A. K.; Feng, T.; Stogryn, E. L.; Purwin, P. E.; Shaw, R. F.; Fishman, C. Appl. Phys. Lett. 1978, 32 (8), 495-497;
CrossRef - Fan, F. R.; Faulkner, L. R. J. Chem. Phys. 1978, 69 (7), 3341-3349.
CrossRef - Tang, C. W. Appl. Phys. Lett. 1986, 48 (2), 183-185.
CrossRef - Yu, G.; Gao, J.; Hummelen, J. C.; Wudl, F.; Heeger, A. J. Science 1995, 270 (5243), 1789-1791.
CrossRef - Roncali, J. Acc. Chem. Res. 2009, 42 (11), 1719-1730.
CrossRef - Liu, Y.; Chen, C.-C.; Hong, Z.; Gao, J.; Yang, Y.; Zhou, H.; Dou, L.; Li, G.; Yang, Y. Sci. Rep. 2013, 3, 3356.
CrossRef - Bundgaard, E.; Krebs, F. C. Sol. Energy Mater. Sol. Cells 2007, 91 (11), 954-985.
CrossRef - Zhang, F.; Wu, D.; Xu, Y.; Feng, X. J. Mater. Chem. 2011, 21 (44), 17590-17600.
CrossRef - Shang, H.; Fan, H.; Liu, Y.; Hu, W.; Li, Y.; Zhan, X. Adv. Mater. 2011, 23 (13), 1554-1557;
CrossRef - Cha, H.; Chung, D. S.; Bae, S. Y.; Lee, M.-J.; An, T. K.; Hwang, J.; Kim, K. H.; Kim, Y.-H.; Choi, D. H.; Park, C. E. Adv. Funct. Mater. 2013, 23 (12), 1556-1565;
CrossRef - Ko, H. M.; Choi, H.; Paek, S.; Kim, K.; Song, K.; Lee, J. K.; Ko, J. J. Mater. Chem. 2011, 21 (20), 7248-7253;
CrossRef - Chen, Y.-H.; Lin, L.-Y.; Lu, C.-W.; Lin, F.; Huang, Z.-Y.; Lin, H.-W.; Wang, P.-H.; Liu, Y.-H.; Wong, K.-T.; Wen, J.; Miller, D. J.; Darling, S. B. J. Am. Chem. Soc. 2012, 134 (33), 13616-13623;
CrossRef - Rousseau, T.; Cravino, A.; Bura, T.; Ulrich, G.; Ziessel, R.; Roncali, J. Chem. Commun. 2009, (13), 1673-1675.
CrossRef - Sonogashira, K. J. Organomet. Chem. 2002, 653 (1–2), 46-49.
CrossRef - Stille, J. K. Angew. Chem. Int. Ed. Engl. 1986, 25 (6), 508-524.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


