Synthesis and Biological Evaluation of Newly Synthesized Flavones
Amreen Khan1* , Anamika Jain1 and Manohar Solanki2
, Anamika Jain1 and Manohar Solanki2
1Department of Chemistry, Govt. Holkar Science College, Indore-452017 (M.P.) India.
2Department of Chemistry, Govt. College, Thandla-457777 (M.P.) India.
Corresponding Author E-mail: khanamrinkhan9@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/400231
Article Received on : 20 Jan 2024
Article Accepted on :
Article Published : 09 Apr 2024
Reviewed by: Dr. Sheetal Jangde
Second Review by: Dr. Reena Gami Patidar
Final Approval by: Dr. Murat HATİPOĞLU
In the present investigation, newly chalcones derivatives of flavonoids (E)-3-(2-chlorophenyl)-1-(2-hydroxyphenyl) prop-2-en-1-one and 2-(2-chlorophenyl)-4H-chromen-4-one synthesized through the Claisen-Schmidt condensation reaction. The initial structure of the newly synthesized compounds have been verified by FT-IR, 1HNMR, and 13C NMR spectroscopic methods. These two flavones were evaluated for their antibacterial activity using the Kuber agar well diffusion method on the bacterium Staphylococcus aureus (S. aureus) and Escherichia coli (E. coli). The results displayed that both compounds have superior antibacterial activity against bacteria, S. aureus and E. coli.
KEYWORDS:Antibacterial activity; Claisen-Schmidt condensation reaction; Chalcone; FT-IR; Flavonoids; 1HNMR;
Download this article as:| Copy the following to cite this article: Khan A, Jain A, Solanki M. Synthesis and Biological Evaluation of Newly Synthesized Flavones. Orient J Chem 2024;40(2). |
| Copy the following to cite this URL: Khan A, Jain A, Solanki M. Synthesis and Biological Evaluation of Newly Synthesized Flavones. Orient J Chem 2024;40(2). Available from: https://bit.ly/43Onpdn |
Introduction
Flavonoids constitute a diverse group comprising over 4000 polyphenolic compounds inherent in plant-derived foods. The primary basis for the systematic classification of these compounds is the saturation level and aperture of the principal pyran ring, which leads to the formation of flavones, flavanols, flavonols, isoflavones, and flavanonols. They share a fundamental phenyl-benzopyrone structure (C6-C3-C6)1,2. Flavones hold a distinctive position within the field of natural and synthetic organic chemistry due to their consequential biological actions, encompassing antioxidant3-7, anxiolytic8, antitumor9-11, anti-inflammatory12-14, bactericidal15, antiulcer, and thrombosis16 effects.
There are several approaches for preparing chalcone, which is a crucial precursor for the synthesis of flavones. Chalcones are usually produced by the Claisen-Schmidt condensation reaction which involves substituted benzaldehyde and acetophenone followed by a strong base ( NaOH, KOH, KOtBu and MgO). Moreover, chalcones can be synthesized through advanced methods such as ultrasonic vibration and microwave irradiation procedures17-19.
Chalcones, represent major alternate metabolites in the flavonoid (Fig.1) class, abundantly present in edible plants. Their radical quenching property, owing to phenolic groups, has spurred research into chalcone-rich plant extracts for therapeutically beneficial compounds. Historically, chalcones like chalconarngenin, phloretin, and phloridzin have been associated with diverse therapeutic applications14-19.
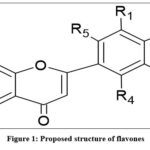 |
Figure 1: Proposed structure of flavones. |
Studies reveal that both natural and synthetic flavonoid and their analogs display a broad spectrum of pharmaceutical actions, including antioxidant, antitubercular, antimicrobial, anti-inflammatory, anti-plasmodial, antiviral, antitumor, antileishmanial, modulation of P-glycoprotein-mediated multi-drug resistance, and immune supportive potential20,21. Interestingly, substances that possess a chalcone-based structure or template also have a significant therapeutic impact on the nervous, cerebrovascular, and cardiovascular systems. In addition to their anti-spasmodic, sedative, anti-thrombic, tranquilizing, analgesic, vasodilatory, anesthetic, anti-coagulating, estrogenic,anti-convulsant, and diuretic qualities.
Furthermore, flavonoid are regarded as vital pharmacophores in numerous bioactive natural commodities, showcasing varied biological actions. Typical examples of naturally existing bioactive chalcones include xanthohumol and cardamonin, with antimutagenic, vasorelaxant, anti-inflammatory, and broad-spectrum tumor chemo-preventive properties22,23. In the current investigation, chalcones are used as the primary precursor for the creation of active flavonoids and their biological evaluation.
Materials and Methods
In the present investigation, we utilized A.R. grade chemicals and reagents (2-Hydroxy acetophenone, 2-chlorobenzaldehyde, and DMSO) obtained from E. Merck. The development of reactions was monitored using thin-layer chromatographic precoated plates (E. Merck). The FT-IR spectra of the compound were recorded from the NICOLET 6700, Thermo Scientific spectrophotometer. The 1HNMR signals were captured at optimal temperature via a 400 MHz liquid-state NMR Spectro-meter in DMSO-d6 (Brüker Biospin), with tetramethyl silane serving as the internal standard.
Antibacterial activity has been determined via the Kuber agar well diffusion technique26. The concentration of the compound for testing is 50 mg/ml, and the test organisms used were E. coli and S. aureus, 8 mm diameter well was made in an agar plate in which the 50 µl compound was loaded, and incubated at 37℃ for 24 hours, next day the plates were observed for the presence of the zone of inhibition.
Extensive mechanism for the synthesis of substituted chalcone
The chalcone of every target has been created by utilizing a standard Claisen-Schmidt condensation24,25 reaction to react equimolar amounts of substituted acetophenone with substituted benzaldehyde. This reaction creates an α, β-unsaturated ketone bridge connecting ring A and ring B of the chalcone basic structure. In the present investigation, the synthesis procedure has been completed in two steps as follows-
Step one
To a mixture comprising 2-hydroxyacetophenone (1 equivalent) and a substituted aromatic aldehyde (2-chlorobenzaldehyde), 10 ml of ethanol was included dropwise with stirring. The mixture was sustained in an ice bath for 15–20 minutes, and freshly prepared 10 ml of 60% NaOH was added dropwise to the above mixture with persistent stirring. The reaction solution was standing for five hours, and the reaction development was scrutinized by thin-layer chromatography. Subsequently, it was acidified with diluted hydrochloric acid, and the precipitate was filtered and washed with cold water, dried, and recrystallized by ethanol.
Yield: 78%
Color: Pale yellow
MP : 144ºC
Molecular Mass: 258.90 g/mol
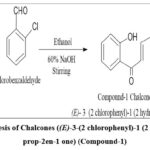 |
Figure 2: Synthesis of Chalcones ((E)-3-(2 chlorophenyl)-1 (2 hydroxyphenyl) prop-2en-1 one) (Compound-1) |
Step two
To equimolar quantities of the compound obtained from the preceding step, DMSO and a catalytic amount of solid iodine were added dropwise. The mixture of chemical reactions were refluxed for about 2 hours and monitored by TLC. It was then poured into crushed ice. The resultant mixture was recrystallized using diethyl ether. The ethereal layer was extracted and dried under a vacuum. The synthetic pathway is presented in fig. 3.
Yield: 76%
Color: Silvery white
MP : 146ºC
Molecular Mass: 256.69 g/mol
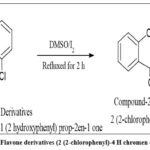 |
Figure 3: Synthesis of Flavone derivatives (2 (2-chlorophenyl)-4 H chromen 4-one.) (Compound-2). |
Results and Discussion
The two flavonoids (Compound-1 and Comound-2) were prepared by the Claisen-Schmidt condensation procedure. Physical analysis suggested that the newly prepared compounds are solids with different colors (Pale yellow and silvery white), and the resultant yield is adequate. Both compounds are soluble in the general organic solvent viz; DMF, DMSO, hexene, chloroform, and benzene, etc.
FT-IR Interpretation of synthesized compounds
Chalcone (Compound-1)
FT-IR spectrum of Chalcone (Compound-1) (fig. 4) is not overly elaborate having characteristic bends. The broad bands at the higher frequency site 3523 cm-1 is revealed the υ(O-H) stretching vibration of the -OH functional group27,28. The stretching frequency υ(C-H) is usually displayed as a weak intensity band with a range of 3035-3050 cm-1 and it appears at 3041 cm-1 as a weak band1. The well-defined bands at 1649 cm-1 is the individual properties of the carbonyl Compound its due to υ(C=O) group. The sharp υ(C=C) band at 1554 cm-1 is owing to the phenyl ring vibration29,30. The medium band at the lower site 767 cm-1 is due to the υ(C-Cl) vibration of the chlorine and bond in between the phenyl ring31.
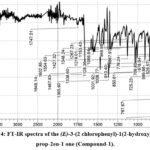 |
Figure 4: FT-IR spectra of the (E)-3-(2 chlorophenyl)-1(2-hydroxyphenyl) prop-2en-1 one (Compound-1). |
Flavone (Compound-2)
FT-IR spectra of Flavone compound-2 (Fig. 5) showed useful information about the newly prepared compound. Aromatic compounds usually displayed several weak singles in the region 3110-3045 cm-1 due to the υ(C-H) stretching32,33. In the IR spectrum, the bend appears at 3041 cm-1 as a weak signal is due to the aromatic C-H group34. The broad band’s at the higher frequency site (3523 cm-1) disappears in the compound-2 is confirmed the oxidation of chalcone compound-1 into flavone compound-2. The well-defined bands at 1639 cm-1 is the individual properties of the carbonyl Compound its due to υ(C=O) group. The medium band at 1571 cm-1 are due to the C-C skeletal mode of vibration of the aromatic ring35-36. The sharp band at 1386 cm-1 is due to the phenyl ring υ(C=C) vibration29-30. The medium band at the lower frequency site 746 cm-1 is due to the chlorophenyl, υ(C-Cl) vibration31.
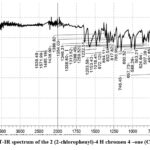 |
Figure 5: FT-IR spectrum of the 2 (2-chlorophenyl)-4 H chromen 4 –one (Compound-2). |
1HNMR and 13CNMR interpretation of newly synthesized compounds
Chalcone (Compound-1)
In the 1HNMR band, a significant chemical shift value appears as a singlet at 8.69 ppm, assigned to the O-H proton, exhibiting high de-shielding due to intra-molecular hydrogen bonding with the carbonyl oxygen atom. A doublet was displayed at 7.89 ppm and 7.67 with huge coupling constant J = 15.5 and 15.5 Hz, respectively37, which are characteristics of trans olefinic protons (α and β). Other signals and the total proton count align precisely with the identified compound’s structure. Despite the synthesized compound having 15 carbons, the 13C NMR spectrum displays thirteen signals, ascribed to the symmetric character of the 4-chlorophenyl ring38.
 |
Figure 6: 1HNMR spectrum of the (E)-3-(2 chlorophenyl)-1(2-hydroxyphenyl) prop-2en-1 one (Compound-1). |
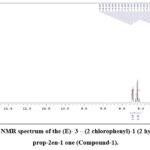 |
Figure 7: 13C NMR spectrum of the (E)- 3 – (2 chlorophenyl)-1 (2 hydroxyphenyl) prop-2en-1 one (Compound-1). |
Flavone (Compound-2)
In the 1H NMR signal, the bend at 8.22–7.55 ppm (doublet) and 7.64–7.43 ppm (double doublet) is due to the ring hydrogen (d, 2H, Ar and dd, 2H, Ar) respectively. The doublet bends at 7.92–7.60 ppm and a double doublet at 7.50–7.47 ppm is due to the chlorophenyl ring (dd, 2H, Ar–Cl and d, 2H, Ar–Cl) respectively. The strong bend at 6.99 ppm is due to the (s, 1H, =CH-) proton present in this compound39. All the aromatic protons are observed in predictable regions. In the 13C NMR signals the aromatic (=CH-) carbon appeared at 108.03 ppm and C-O carbon appeared at 155.85 and 157.45. The bend at 177.52 is due to the (C=O) carbon. Newly synthesized compound having 15 carbons, the 13C NMR spectrum displays thirteen signals, ascribed to the symmetric character of the 4-chlorophenyl ring38. The 13C NMR signals offer direct evidence about the carbon skeleton of the newly synthesized compound.
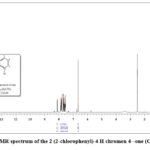 |
Figure 8: 1HNMR spectrum of the 2 (2-chlorophenyl)-4 H chromen 4 –one (Compound-2). |
 |
Figure 9: 13C NMR spectrum of the 2 (2-chlorophenyl)-4 H chromen 4 –one (Compound-2). |
Biological Evaluation
The antibacterial action of the two newly produced compounds, (compound-1 and compound-2), were tested against G– microbe (E. coli) and G+ microbe (S. aureus) by the agar well diffusion procedure. The results revealed that the synthesized compounds exhibited superior antimicrobial activity against G+ microbe S. aureus, with zones of inhibition measuring 28 mm and 25 mm for compound-1 and compound-2, respectively. Conversely, in the case of G– microbe E. coli, the antibacterial activity was observed to be 22 mm and 19 mm for compound-1 and compound-2, respectively (Fig.10).
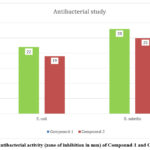 |
Figure 10: Antibacterial activity (zone of inhibition in mm) of Compound-1 and Compound-2. |
Conclusion
Synthesis of flavonoids was effectively completed through a condensation reaction and the purity of compounds were determined using melting point and TLC measurements. In order to synthesized compound further confirmed by the FT-IR, 1HNMR, and 13C NMR spectral studies. Antibacterial activity of the synthesized, Compound-1 and Compound-2, demonstrated notable activity against G+ microbe (S. aureus) and G– microbe (E. coli). Specifically, superior antibacterial efficacy was observed against S. aureus, emphasizing the potential significance of these synthesized flavonoids in combating bacterial infections. The comprehensive synthesis and characterization, coupled with promising antibacterial properties, underscore the utility of the developed compounds for further exploration in pharmaceutical and medicinal applications.
Acknowledgment
We would like to thank IIT Indore for the spectral analysis and the head of the microbiology department at Govt. Holkar Science College, Indore, for their assistance with the biological studies.
Conflict of Interest
According to the authors, there are no conflicts of interest associated with this work’s publishing.
References
- Middleton, E.; Kandaswami, C. “The Flavonoids Advances in Research” Chapman and Hall, London, 1986, 619-652.
- Harborne, J. B.; Williams, C. A. Phytochemistry, 2000, 55(6), 481-504.
CrossRef - Pal, D.; Verma, P. Int. J. Pharm. Pharm. Sci., 2013, 5(3), 95-98.
CrossRef - Rice-Evans, C. A.; Miller, N. J.; Paganga, G. Free Radic. Biol. Med., 1996, 20(7), 933-956.
CrossRef - Rice-Evans, C. Curr. Med. Chem., 2001, 8(7), 797-807.
CrossRef - Pietta, P. G. J. Nat. Prod., 2000, 63(7), 1035-1042.
CrossRef - Chan, E. C.; Pannangpetch, P.; Woodman, O. L. J. Card. Pharm., 2000, 35(2), 326-333.
CrossRef - De Almeida, E. R.; Xavier, H S.; Chaves, T. M.; Couto, G. B.; Aragao-Neto, A. C.; Silva, A. R. Int. J. Appl. Res. Nat. Prod., 2009, 2(4), 44-51.
- Liu, Y. L.; Ho, D. K.; Cassady, J. M.; Cook, V. M.; Baird, W. M. J. Nat. Prod., 1992, 55(3), 357-363.
CrossRef - Ramos, S. J. Nut. Biochem., 2007, 18(7), 427-442.
CrossRef - Ren, W.; Qiao, Z.; Wang, H.; Zhu, L.; Zhang, L. Med. Res. Rev., 2003, 23(4), 519-534.
CrossRef - Shin, J. S.; Kim, K. S.; Kim, M. B.; Jeong, J. H.; Kim, B. K. Bioorg. Med. Chem. Lett., 1999, 9, 869-874.
CrossRef - Dao, T. T.; Chi, Y. S.; Kim, J.; Kim, H. P.; Kim, S.; Park, H. Bioorg. Med. Chem. Lett., 2004, 14(5), 1165-1167.
CrossRef - Theja, D. N.; Choudary, T. P.; Reddy, M. I.; Gupta, A.; Reddy, K. U. Int. J. Pharm. Pharm. Sci., 2011, 3(2), 51-54.
- Mostahar, S.; Alam, S.; Islam, A. Ind. J. Chem., 2006, 45B, 1478-1486.
- Tapas, A. R.; Sakarkar, D. M.; Kakde, R. B. Trop. J. Pharm. Res., 2008, 7(3), 1089-1099.
CrossRef - Kakati, D.; Sarma, J. C. Chem Cent. J., 2011, 5(8), 1-5.
CrossRef - Sashidhara, K. V.; Rosaiah, J. N.; Kumar, A. Synth. Commun., 2009, 39(3), 2288-2296.
CrossRef - Li, J. T.; Yang, W. Z.; Wang, S. X.; Li, S. H.; Li, S. H. Ultrason. Sonochem., 2002, 9(5), 237-239.
CrossRef - Ouyang, Y. Li, J.; Chen, X., Fu, X.; Sun, S.; Wu, Q. Biomol., 2021, 11, 894.
CrossRef - Gomes, M. N.; Muratov, E. N.; Pereira, M.; Peixoto, J. C.; Rosseto, L. P.; Cravo, P. V., Neves, B. J. Molecules, 2017, 22, 1210.
CrossRef - Khan, J.; Deb, P. K.; Piya, S.; Medina, K. D.; Devi, R.; Walode, S. G.; Rudrapal, M. Molecules, 2021, 26, 4021.
CrossRef - Martinez, R. M.; Pinho-Ribeiro, F. A.; Steffen, V. S.; Caviglione, C. V.; Fattori, V.; Bussmann, A. J.; Baracat, M. M. Photochem. Photobio. Sci., 2017, 16, 1162–1173.
CrossRef - Dong, F. Jian.; Cheng Z.; Fei K. G.; Zuliang, L. Catal. Commun. 2008, 9, 1924-1927.
CrossRef - Guida, A.; Lhouty, M. H.; Tichit, D.; Figueras, F.; Geneste, P. Appl. Catal. Gen., 1997, 164(1-2), 251-264.
CrossRef - Pandey, A.; Chandel, E. World J. Pharm. Pharm. Sci., 2014, 3(11), 879-898.
- Z. H. Ma, H. B.; Han, Z. B.; Zhou, J. N. J. Mol. Cat., 2009, 1, 46-53.
CrossRef - Bellamy, L. J. The infrared Spectra of Complex Molecules,Chapman and Hall London, 1975, 1.
CrossRef - Chandra S.; Sharma A. K. Spectrochim. Acta A, 2009, 72, 851.
CrossRef - Zati-Hanani S.; Adnan R. C. Sipaut, Sains Malaysiana, 2011, 40(90), 999-1006.
- Mooney E. F. Spectrochimica Acta. 1964, 20,1343-1348.
CrossRef - Wang Y.; Saebo S. J. Mol. Struc., 1993, 281(2-3), 91-98.
CrossRef - Ansari A. M.; Karabacak M.; Kurt M. Spectrochim Acta A, 2011, 82, 444-455.
CrossRef - Silverstein R. M.; Webster F. X. Spectrometric identification of organic compounds John Wiley, Sons, Inc, New York, 1996, 260-286.
- Saxena A.; Saxena R, Orient. J. Chem., 2013,29, 89-595.
CrossRef - Uruş S.; Karabo M.; Koksal, H.; Appl. Organo. Chem. 2018, 32(1), 4022-4031.
- Velayutham S. V.; Chithathoor V. M.; Ramalingam B.; Natarajan V.; Bioint. Res. App. Chem. 2022, 12(6), 7159-7176.
CrossRef - Bunu S. J.; Awala E. V.; Eboh D. D.; Saudi J. Med. Pharm. Sci. 2020, 6(4), 379-38
- Patel S.; Shah U.; Asian J. Pharm. Clin. Res. 2017, 10(2), 403-406
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


