Electro-organic Reactions: Direct and Indirect Electrolysis
Sulekha Sharma*
Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhauri, Bhopal, Madhya Pradesh, India.
Corresponding Author E-mail: sulekhasharma4006@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/400202
Article Received on : 10 Jan 2024
Article Accepted on :
Article Published : 29 Mar 2024
Reviewed by: Dr. Sandhya Kapurwan
Second Review by: Dr. Jyoti Tanwar
Final Approval by: Dr. Dinesh Chand Sharma
Electro-organic synthesis is a new methodology for synthesizing organic molecules, which helped overcome the use of toxic oxidants and expensive catalysts and make the reaction greener. The fundamental concepts of electrochemistry involve simple oxidation and reduction reactions, where electrons act as the greener oxidant and reductant. This review discusses electrochemical principles and basic terminologies that find their roots in physical organic chemistry to influence a spectrum of organic and inorganic reactions. This review also discusses the different modes of electrolysis, i.e., direct and indirect. Finally, the review highlights the importance of direct and indirect electrolysis for various reactions.
KEYWORDS:Direct electrolysis; Electro-organic synthesis; Green reaction; Indirect electrolysis
Download this article as:| Copy the following to cite this article: Sharma S. Electro-organic Reactions: Direct and Indirect Electrolysis. Orient J Chem 2024;40(2). |
| Copy the following to cite this URL: Sharma S. Electro-organic Reactions: Direct and Indirect Electrolysis. Orient J Chem 2024;40(2). Available from: https://bit.ly/3vyqSjw |
Introduction
The depletion of fossil fuels has prompted a growing need for sustainable methods in synthetic applications, leading to the emergence of various techniques, such as photochemistry and electrochemistry.1,2 Electrochemical synthesis has gained considerable popularity among these due to its mild reaction conditions and excellent functional group tolerance.3-7 The roots of electrochemical methodology trace back to Faraday’s introduction in 1830, aiming to drive non-spontaneous organic reactions using current.8 Despite early contributions from Faraday, Kolbe, Haber, and others, electrochemistry took nearly two centuries to become mainstream in organic chemistry.3 ,8-15
The slow adoption of electrochemical methodology can be attributed to the unfamiliarity of organic chemists with the experimental setup and electrochemical techniques, resulting in challenges in reproducing results.16 However, the 21st century witnessed a significant surge in the field, fueled by the commercialization of standard electrolytic cells and equipment designed for synthetic purposes.4, 5, 16-18 Recent advancements in electrochemical synthesis primarily stem from the desire to replace super stoichiometric, harmful, and expensive chemical oxidants/reductants, focusing on minimizing waste by-products. Additionally, the shift towards electricity as a cheaper source, derived from sustainable and renewable energy sources rather than fossil fuels, 19-21 has played a crucial role in the field’s growth.
Electro-organic synthesis, as a form of electrochemical methodology, employs electricity as a reagent for constructing organic molecules. By harnessing the electrode potential to oxidize/reduce a species,22 electrochemical routes offer sustainable, safe, atom-economic, inherently green, and powerful eco-friendly tools for organic synthesis. This approach contributes to environmental sustainability and enhances our understanding of chemical reactions and their mechanisms. 7, 20, 23-25
Concise History of Electro-Organic Synthesis
This segment provides a concise overview of the research area, spanning its origins, historical development, and progress. The rich history of utilizing electric current to synthesize organic molecules can be traced back to the ground-breaking contributions of Faraday and Kolbe during the 19th century. 3, 9, 26 In 1849, Kolbe notably elucidated the synthesis of alkanes through the decarboxylative dimerization of two carboxylic acids (Scheme 1).9
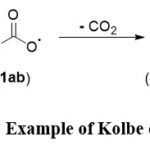 |
Scheme 1: Example of Kolbe electrolysis. |
Subsequently, in 1898, Haber conducted a significant experiment involving the electrolytic reduction of nitrobenzene, resulting in the selective production of aniline and phenylhydroxylamine (Scheme 2). Notably, he observed that phenylhydroxylamine was preferentially formed at a less negative potential, where it consumed 4 electrons per molecule of nitrobenzene. In contrast, a more negative potential led to the formation of aniline, involving the consumption of 6 electrons (Scheme 2). This pivotal discovery played a crucial role in advancing the field further. The experiments underscored the essential role of electrode potential as a fundamental parameter in determining the Gibbs energy value of the electrode process, influencing the selectivity and outcome of the reaction.27
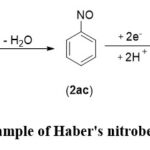 |
Scheme 2: Example of Haber’s nitrobenzene reduction. |
A groundbreaking advancement in electro-organic synthesis occurred with Hickling’s introduction of the potentiostat in 1942. This innovative device facilitated reactions at a constant potential, employing a setup that contained three electrodes where the potential was controlled on the working electrode with respect to the reference electrode. This marked the end of the reliance on galvanostatic conditions in electro-organic synthesis, where organic molecules were charged to a constant current electrolysis, where with time the potential of the reaction keep on rising.3,28 Additionally, the invention of cyclic voltammetry by Randles in 1948 provided a means for the precise measurement of redox potentials of specific functional groups, establishing itself as a crucial tool for electrochemists to this day.3,29 Some interesting classic examples include the Monsanto adiponitrile process, which showcased the scalability of electro-synthetic reactions at industrial scales (Scheme 3).30
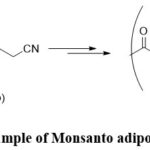 |
Scheme 3: Example of Monsanto adiponitrile process. |
Additionally, Shono and collaborators pioneered the inaugural direct anodic oxidation of the α–methylene group, resulting in the generation of an amide (carbamate) and the formation of a carbon-carbon bond through an N-acyliminium ion intermediate (Scheme 4).3, 31, 32
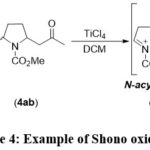 |
Scheme 4: Example of Shono oxidation. |
The evolution of electro-organic reactions continued through pioneering studies, underscoring the pivotal role of electrodes in facilitating organic reactions. The recognition of direct electron transfers between the substrate and the electrode became a well-established concept among scientists. While in the direct electrolysis electron transfer between electrode surface and substrate takes place heterogeneously, these processes often encounter high kinetic barriers in the reactions.33 Additionally, the accumulation of highly charged intermediates near the electrode surface forms an electrical double layer.34-36 Consequently, there is a risk of these intermediates diffusing back into the solution, potentially impacting the chemical reactions of interest.
On the contrary, the remaining intermediate undergoes decomposition at the electrode, leading to electrode passivation. To address the issue of electrode passivation, indirect electrolysis involving an inorganic mediator was introduced in the early 1900s.36 In this approach, a mediator, often a redox catalyst, where heterogeneous electron transfer is replaced with homogeneous electron transfer. Subsequently, the field witnessed a surge in popularity, thanks to the notable contributions of Steckhan. One noteworthy example is the oxidation reaction of benzene to phenol using iron salts, where the presumed “in-situ” formation of Fenton’s reagent takes place (Scheme 5).12, 36, 37
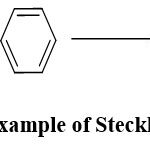 |
Scheme 5: Example of Steckhan reaction. |
Subsequently, in the latter part of the 20th century, Yoshida introduced the concept of electro-auxiliaries. This involves incorporating functional groups such as silicon and sulfur into the substrate to lower their electrochemical potentials. This strategic modification enables enhanced control over regioselectivities and chemoselectivities in the electrochemical reactions (Scheme 6).38, 39
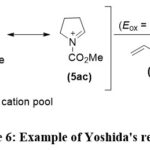 |
Scheme 6: Example of Yoshida’s reaction. |
Significant strides in electro-organic synthesis were propelled by the groundbreaking contributions of esteemed groups, including but not limited to, Little et al.,14, 40 Schafer et al.,41 Lund et al.,42 Moeller et al.,35 Amatore et al.,43 Jutand et al.,43 and Yoshida et al.13 These pioneering works from the early stages laid the foundation for the widespread recognition and popularity of electro-organic chemistry in the 21st century.44-47
Fundamentals of Electro-Organic Synthesis
Electro-organic reactions encompass two distinct processes: the electrode process and the chemical process (Figure 1). In the electrode process, a heterogeneous transfer of electrons transpires, generating a reactive intermediate on the electrode surface. Initially, the reaction is subjected to the necessary electric potential, which results in electrical double layer formation at an electrode-electrolyte interface. Various phenomena, such as diffusion, convection, or migration, come into play at this interface, transforming the reactive species into the active species.48
In the subsequent chemical process, the reactive intermediate formed on the electrode surface diffuses back into the solution, where the desired chemical reaction takes place. Follow-up reactions may involve processes such as elimination, rearrangement, recombination, or substitution, among others.
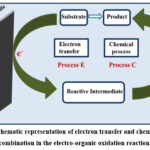 |
Figure 1: Schematic representation of electron transfer and chemical process combination in the electro-organic oxidation reaction. |
Redox process at electrode/electrolyte process
In electrochemical reactions, the redox process involves electron transfer between the substrate and electrode, achieved by controlling potential of the working electrode in relation to the reference electrode. Applying a positive potential to electrode, Fermi level energy of electrode get lowered. Consequently, the energy level of the electrode aligns with highest occupied molecular orbital (HOMO) energy of substrate (Figure 2). This alignment facilitates electron transfer from the substrate to the electrode, a process known as oxidation.49
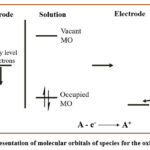 |
Figure 2: Representation of molecular orbitals of species for the oxidation process. |
Likewise, applying a negative potential at the working electrode results in an elevation of the energy of the Fermi level of the electrode. This change aligns the energy level of the electrode with substrates lowest unoccupied molecular orbital (LUMO) energy (Figure 3). Consequently, transfer of electrons occur from surface of electrode to species in solution, and process known as reduction.49
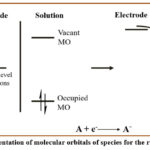 |
Figure 3: Representation of molecular orbitals of species for the reduction process. |
Basic Terminology in Electrochemistry
Charge is a fundamental property of matter, defining its ability to experience a force when situated in an electric or magnetic field. It manifests as either positive or negative, commonly associated with protons or electrons, respectively. In electrochemical reactions, the transfer of electrons is prevalent, and this flow transpires only when the circuit is complete. The principle of conservation of mass and charge dictates that the number of donated electrons must precisely match the number of accepted electrons.50 The calculation of charge is expressed by equation 1.

Herein, Q (C), n, F (C mol-1), and N (mol) represent the charge, number of electrons transferred, Faraday’s constant, and the number of moles of species, respectively.
Voltage refers to the work done to move a test charge from one point to another within a circuit. In an electrochemical system, force is needed to propel electrons from the anode to the cathode. This relationship is represented by equation 2, where V (V) signifies voltage and W (J) denotes work.50
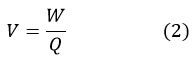
Current quantifies the flow of charge particles per unit of time. Typically, these particles are electrons moving through a wire, although, in electrolytic solutions, the charge carriers are ions that facilitate the circuit’s completion. Another interpretation of current is the flow of one coulomb of charge in one second. This is mathematically represented by equation 3, where I (A) denotes current, and t (s) signifies time.50
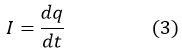
Current density is characterized as the quantity of current passing through per unit area. In electrochemistry, it holds greater significance as this term becomes independent of the electrode surface area. In contrast, the current quantity is proportional to the electrode surface area. The relationship expresses the current density calculation in equation 4, where j (A cm⁻²) signifies current density and A (cm²) represents the electrode area.50

Faradaic process involves the transfer of electrons across the electrode-electrolyte interface, leading to the oxidation or reduction of a species. In these reactions, the number of chemical transformations resulting from the current flow is directly proportional to the amount of electricity passed. These reactions adhere to Faraday’s law and are known as faradaic processes.50
Non-faradaic process occurs when no charge-transfer reaction occurs across the electrode-electrolyte interface within a specific range of potentials. In this range, the reactions are both thermodynamically and kinetically unfavourable. Therefore, currents associated with non-faradaic processes depend on the electrode surface area and the electrolyte concentrations.50
Mass transfer refers to the speed at which the redox material moves from the solution to the electrode surface. This movement is driven by disparities in either electrical or chemical potentials between the electrode-electrolyte interface and the bulk solution.
There are various modes of mass transfer, such as:50
Migration
The transport of charged particles due to the influence of the electric field It is a gradient of electric potential.
Diffusion
The transportation of charge material along a concentration gradient represented in Figure 4.
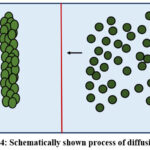 |
Figure 4: Schematically shown process of diffusion. |
Convection
The conveyance of charge material through stirring (hydrodynamic transport) illustrated in Figure 5.
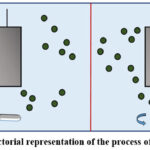 |
Figure 5: Pictorial representation of the process of convection. |
Modes of Electrolysis
Direct electrolysis
In the direct mode of electrolysis, electron transfer occurs heterogeneously across the electrode-electrolyte interface, simplifying the determination of the redox potential of the substrate in an electrochemical reaction. This direct electron transfer mechanism involves a few steps. In the initial step, the substrate migrates towards the electrode surface, where it undergoes adsorption onto the electrode surface. Subsequently, in the following step, electron transfer occurs between the substrate and the electrode, resulting in the formation of a reactive intermediate.
In the final step, the generated reactive intermediate undergoes a chemical reaction, leading to either the desorption of the formed product or the desorption of the reactive intermediate, followed by its transformation into the final product, as exhibited in Figure 6.33,36
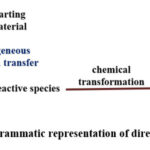 |
Figure 6. Diagrammatic representation of direct electrolysis. |
Therefore, by leveraging the fundamental principles of electrochemistry and significant breakthrough in electrochemical for organic compounds synthesis using direct electrolysis have been noticed in recent years. Notably, there has been a substantial surge in metal-free functionalizations. The latest contributions from various research groups, including Baran et al.,44 Waldvogel et al.,51 Lei et al.,45 and others,46 are discussed below.
Examples of direct electrolysis
Electrochemical C-H Amination
Yoshida et al. have introduced a novel methodology utilising electrochemistry, where the electro-oxidation of aromatic compounds occurs when pyridine is also used in the system. This leads to a subsequent formation of N-aryl pyridinium ions (Scheme 7). The electrochemical reaction was executed in an undivided cell employing a mixture of pyridine/acetonitrile as the solvent system and 0.3 M tetrabutylammonium tetrafluoroborate (TBABF₄) as the supporting electrolyte.52
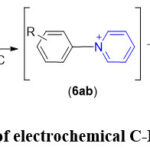 |
Scheme 7: Example of electrochemical C-H amination reaction. |
Electro-Oxidative Reaction of Aminophenols and Benzenesulfonate:
Little et al. explored the anodic oxidation of aminophenols through cyclic voltammetry, revealing its capability for a Michael addition reaction. Notably, the benzenesulfonate group was introduced selectively on an amino group at its meta position (Scheme 8). The electrochemical anodic oxidation reaction was performed by applying a potential of 0.4 V with respect to Ag wire in presence of a sodium acetate/acetic acid buffer.53
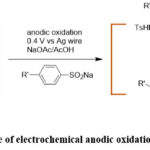 |
Scheme 8: Example of electrochemical anodic oxidation of aminophenols. |
Electrochemical Oxidative N-N Dimerization
Baran et al. have introduced a distinctive electrochemical methodology for the N-N dimerization and it has been utilized on carbazoles and carbolines. This methodology was particularly valuable for the synthesis of dixiamycin B from xiamycin A, addressing the challenge of obtaining N-N-linked natural products. The reaction was conducted at a potential of 1.15 V in a simple undivided cell setup. A carbon electrode served as the anode, and tetraethylammonium bromide (Et₄NBr) functioned as the electrolyte (Scheme 9).54
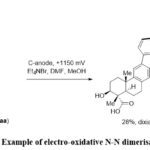 |
Scheme 9: Example of electro-oxidative N-N dimerisation. |
Dehydrogenative Coupling of Phenols and C-C bond formation
Waldvogel et al. have demonstrated metal-free electrochemical oxidative reaction of phenols with various electron withdrawing groups, which give rise to carbon-carbon bonds, as illustrated in Scheme 10. These reactions are straightforward to conduct under electrochemical conditions and exhibit scalability. Importantly, this reaction condition aligns with environmental considerations by minimizing the production of waste by-products.55
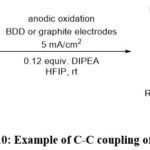 |
Scheme 10: Example of C-C coupling of phenols. |
Despite these advances, electrochemical metal-free C(sp3)-C(sp3) bond formation is still very less explored due to its complexity of bond formation. One of the reports includes:
Semipinacol Rearrangements of Allylic Alcohols Using Electrochemistry to Construct All-Carbon Quaternary Stereocenters:
Zhang et al. have documented a pioneering instance of electrochemical trifluoromethylation and sulfonylation of allylic alcohols via semipinacol rearrangement (Scheme 11). Using stable and cost-effective sodium sulfinates (RSO₂Na) as reagents is notable. Furthermore, this electrochemical reaction avoids use of transition metal catalysts, stoichiometric oxidants, as well as harsh conditions.56
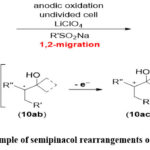 |
Scheme 11: Example of semipinacol rearrangements of allylic alcohols. |
Indirect electrolysis
Direct electrolysis is a specific scenario within electrochemical reactions where electron transfer at electrode is a heterogeneous process. In contrast, indirect electrolysis shifts the electron transfer step to homogeneous process, creating the electrochemical active specie which acts as mediator or electrocatalyst. This mediator constitutes redox couple which is reversible in nature and formed at surface of electrode, which accelerates reaction of interest. To carry out reaction catalytic amount of mediator is needed, thereby allowing the avoidance of reagent waste generation. Additionally, it serves to prevent electrode passivation, eliminating kinetic inhibition.12, 33, 36
Principle of indirect electrolysis
The underlying principle of indirect electrolysis involves substituting the step of heterogeneous electron transfer with a homogeneous electron transfer (Figure 7). Electrochemically the electroactive species can be generated and regenerated in situ within the same electrolytic setup and this is termed as in cell process and when it takes place in different cell, it is known as ex cell process. In the former process, a redox reagent is employed in small amounts (catalytic) as they can be continuously recreated on electrode. Consequently, the reaction conditions are carefully selected to ensure that only the mediator performs electron transfer with electrode. At the same time, substrate, intermediate, and product should not interfere with catalysts when they regenerate. Additionally, during in cell process, activated form of the mediator must exhibit sufficient stability to react promptly with the substrate of interest. Conversely, the ex-cell process mediator should be stable enough for transfer to a new vessel. Therefore, the in-cell approach is generally preferred for economic reasons. 12, 33, 36
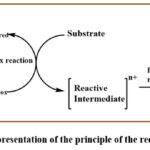 |
Figure 7: Representation of the principle of the redox mediator. |
Mechanism of indirect electrolysis
Indirect electrolysis consists of two types of electron transfer mechanisms.36
Outer sphere mechanism
Inner sphere mechanism
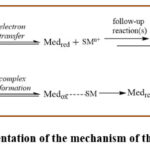 |
Figure 8: Representation of the mechanism of the redox mediator. |
In the outer sphere mechanism, outer sphere electron transfer or non-bonded transfer of electron occurs amidst mediator and the substrate (Figure 9). Conversely, in the inner sphere reaction pathway, formation of bond occurs in-between active form of mediator and substrate due to a homogeneous chemical reaction amongst two species. Inner sphere electron transfer occurs through the charge transfer complex (Figure 9). Notably, reactions following this mechanism can be conducted at much lower potentials (>1 Volt) from actual potential of substrate itself. 36
Advantages of indirect electrolysis
Indirect electrolysis offers several advantages over direct electrolysis, including:36
Elimination of kinetic inhibition. This results in reduced overpotential and an acceleration of the reaction kinetics.
Prevention of electrode passivation by avoiding direct electron transfer.
The ability to conduct electrochemical reactions at a much lower potential of substrate. Consequently, mild conditions can be used for reactions, minimizing side reactions.
Improved yield and selectivity of reactions.
Choice of a suitable mediator
To be a suitable mediator for an electrochemical reaction, certain essential criteria should be met: 36
The mediator should have lower redox potential than substrate. However, the potential difference between these two species should not be so significant that the reaction becomes practically impossible. As a general guideline, the outer-sphere mechanism is expected not to exceed 0.5 V, while the inner-sphere mechanism can tolerate up to 2 V.
There should be a reversible and rapid electron transfer amid mediator and surface of electrode, and also between reactant and mediator. Or else, increasing the surface area of the electrode or elevating the reaction temperature enhance slow rate of reaction.
The mediator in its active form should not react toward other species to avoid unwanted reactions.
The mediator should exhibit high solubility in the solvent to form a homogeneous solution. Alternatively, a biphasic electrolyte system or co-solvent can be considered.
Technique to evaluate the electrocatalytic process
Cyclic voltammetry proves to be a valuable technique for assessing electrocatalytic processes. It aids in determining the difference in potentials of the catalyst as well as reacting specie, facilitating identification of the catalytic activity onset. The efficiency in catalytic process is calculated by measure of current response in a cyclic voltammogram at the mediator’s potential when both the mediator and substrate are present in the solution. Consequently, efficient electron transfer processes result in the appearance of a catalytic current. Figure 9 illustrates typical cyclic voltammetry responses. The black dashed line represents the voltammogram of only the mediator, while the addition of a substantial amount of substrate leads to an increase in peak current (red dotted line). The introduction of a base further enhances the catalytic current (blue dash-dotted line), primarily due to a significant acceleration in the deprotonation step rate. 33, 36
 |
Figure 9: Representation of the effect of base and mediator on the cyclic voltammogram. |
2,2,6,6-Tetramethylpiperidine-N-oxyl Radical (TEMPO) Mediated Electrosynthesis
Concepts of indirect electrolysis for organic synthesis was first introduced by Steckhan in the 1980s.12,57 A comprehensive survey of this field was later conducted by Little and Francke36 in 2014. In recent years, Stahl and colleagues have made significant contributions, extensively utilising TEMPO and its derivatives as mediators for the electro-oxidation of benzyl alcohols. 58-62 Furthermore, various research groups, including Xu et al., 63 Lin et al., 64 Mei et al., 65 and others, have expanded applications in TEMPO catalyzed reactions under electrochemical conditions.
Examples of these contributions include:
Electrochemical Oxidation of Aldehydes and Alcohols to Carboxylic Acids using 4-Acetamido TEMPO
Stahl and colleagues introduced an electrocatalytic method for the oxidation of alcohols and aldehydes utilizing the mediator 4-acetamido-2,2,6,6-tetramethyl-piperidinedin-1-oxyl (ACT) to their respective acids, Scheme 12.60 The reactions were conducted at room temperature using a cost-effective catalyst in an aqueous medium where carbonate/bicarbonate buffer served as both the base and electrolyte.
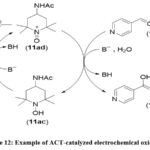 |
Scheme 12: Example of ACT-catalyzed electrochemical oxidation. |
Electrochemical C-H Thiolation Reaction using TEMPO catalysis
Xu et al. devised a reagent- and metal-free synthesis approach for thiazolopyridines and benzothiazoles using TEMPO, as illustrated in Scheme 13. 63 They carried out the reaction using high surface area RVC electrode and platinum cathode and also delved into the reaction mechanism and found that the reaction approaches via inner-sphere electron transfer.
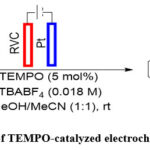 |
Scheme 13: Example of TEMPO-catalyzed electrochemical C-H thiolation. |
TEMPO−N3 Charge-Transfer Complex Mediated Azidooxygenation of Alkenes using Electrochemistry
Lin and colleagues discovered an effective and gentle electrochemical method for accessing diverse vicinal C-N along with C-O difunctionalization products usin alkenes, utilizing TEMPO as the mediator (Scheme 14). 64 They conducted in-depth studies on the reaction mechanism and determined that the one-electron oxidation of TEMPO produces electrochemically oxoammonium ions. Subsequently, the oxoammonium ion forms a charge-transfer complex with azide TEMPO-N3 i.e. the charge transfer complex to generate an azidyl radical (Scheme 14).
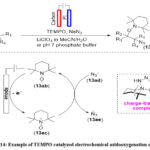 |
Scheme 14: Example of TEMPO-catalyzed electrochemical azidooxygenation of alkenes. |
Electrochemical TEMPO-Catalyzed Enantioselective Oxidative Reactions of Secondary Acyclic Amines
The reaction of accylic amines was demonstrated by Mei and colleagues and they reported asymmetric coupling using electrochemistry where the two platinum electrodes were used as the anode and cathode. The introduction of an N-oxyl radical as a redox mediator enables the selective oxidation of the substrate over the product, despite a slight difference in their oxidation potentials of approximately 13 mV. This electrochemical process occurs without the need for stoichiometric additives such as metals, oxidants, and electrolytes, ensuring excellent compatibility with various functional groups. The mechanistic investigations indicate that proton-mediated racemization of the product is averted through the reduction of protons at the cathode. The notable achievements in this research were enantioselectivity and diastereoselectivity, which resulted in good to excellent yields (Scheme 15). 65
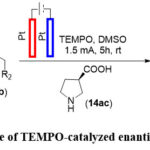 |
Scheme 15: Example of TEMPO-catalyzed enantioselective coupling. |
Conclusion
This review comprehensively discusses the basic concepts of electrochemistry and various driving factors behind electrochemical reactions. It summarises diverse efforts in making various electrochemical organic reactions greener. Throughout the years, the electrochemical approach has proven effective in oxidizing and reducing a wide range of substrates only by using electrodes as the source of oxidant/reductant. This versatile methodology has been applied in various reactions, i.e. direct/indirect electrolysis using a simple electrochemical setup. The breadth of chemistry explored through electrochemical studies makes it a unique and invaluable approach, difficult for other methods to match.
Acknowledgement
This research has received no dedicated funding from any sources.
Conflict of interest
The author declares no conflict of interest.
References
References
- Pollok, D.; Waldvogel, S. R. Chem. Sci. 2020, 11, 12386-12400.
CrossRef - Cernansky, R. Nature 2015, 519, 379-380.
CrossRef - Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230-13319.
CrossRef - Waldvogel, S. R.; Lips, S.; Selt, M.; Riehl, B.; Kampf, C. J. Chem. Rev. 2018, 118, 6706-6765.
CrossRef - Horn, E. J.; Rosen, B. R.; Baran, P. S. ACS Cent. Sci. 2016, 2, 302-308.
CrossRef - Kärkäs, M. D. Chem. Soc. Rev. 2018, 47, 5786-5865.
CrossRef - Wiebe, A.; Gieshoff, T.; Möhle, S.; Rodrigo, E.; Zirbes, M.; Waldvogel, S. R. Angew. Chem. Int. Ed. 2018, 57, 5594-5619.
CrossRef - Lund, H. J. Electrochem. Soc. 2002, 149, S21.
CrossRef - Kolbe, H. J. Prakt. Chem. 1847, 41, 137-139.
CrossRef - Faraday, M. Ann. Phys. 1834, 109, 433-451.
CrossRef - Vijh, A. K. Chem. Rev. 1967, 67, 623-664.
CrossRef - Steckhan, E. Angew. Chem. Int. Ed. Engl. 1986, 25, 683-701.
CrossRef - Yoshida, J.-i.; Murata, T.; Isoe, S. Tetrahedron Lett. 1986, 27, 3373-3376.
CrossRef - Little, R. D.; Schwaebe, M. K. Electrochemistry VI Electroorganic Synthesis: Bond Formation at Anode and Cathode, Steckhan; Springer Berlin Heidelberg: Berlin, Heidelberg, 1997.
- Yoshida, J.-I.; Suga, S.; Suzuki, S.; Kinomura, N.; Yamamoto, A.; Fujiwara, K. J. Am. Chem. Soc. 1999, 121, 9546-9549.
CrossRef - Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Int. Ed. 2018, 57, 4149-4155.
CrossRef - Fuchigami, T.; Atobe, M.; Inagi, S., Fundamentals and applications of organic electrochemistry: synthesis, materials, devices; John Wiley & Sons: 2014.
CrossRef - Luca, O. R.; Gustafson, J. L.; Maddox, S. M.; Fenwick, A. Q.; Smith, D. C. Org. Chem. Front. 2015, 2, 823-848.
CrossRef - Bairagi, A. S. Curr. Res. Green Sustain. Chem. 2022, 5, 100313.
CrossRef - Frontana-Uribe, B. A.; Little, R. D.; Ibanez, J. G.; Palma, A.; Vasquez-Medrano, R. Green Chem. 2010, 12, 2099-2119.
CrossRef - Devi, S.; Jyoti; Kiran; Wadhwa, D.; Sindhu, J. Org. Biomol. Chem. 2022, 20, 5163-5229.
CrossRef - Cardoso, D. S. P.; Šljukić, B.; Santos, D. M. F.; Sequeira, C. A. C. Org. Process Res. Dev. 2017, 21, 1213-1226.
CrossRef - Zhu, C.; Ang, N. W. J.; Meyer, T. H.; Qiu, Y.; Ackermann, L. ACS Cent. Sci. 2021, 7, 415-431.
CrossRef - Rodrigues, R. M.; Thadathil, D. A.; Ponmudi, K.; George, A.; Varghese, A. ChemistrySelect 2022, 7, e202200081.
CrossRef - Ali, T.; Wang, H.; Iqbal, W.; Bashir, T.; Shah, R.; Hu, Y. Adv. Sci. 2023, 10, 2205077.
CrossRef - Faraday, M. Ann. Phys. 1834, 109, 481-520.
CrossRef - Liu, Q.; Tadrent, S.; Proust, C.; Gomez, F.; Khelfa, A.; Luart, D.; Len, C. Chem. Eng. Sci. 2020, 211, 115275.
CrossRef - Hickling, A. Trans. Faraday Soc. 1942, 38, 27-33.
CrossRef - Randles, J. E. Trans. Faraday Soc. 1948, 44, 327-338.
CrossRef - Baizer, M. M. Naturwissenschaften 1969, 56, 405-409.
CrossRef - Jones, A. M.; Banks, C. E. Beilstein J. Org. Chem. 2014, 10, 3056-72.
CrossRef - Onomura, O.2012, 85, 2111.
CrossRef - Hilt, G. ChemElectroChem 2020, 7, 395-405.
CrossRef - Sperry, J. B.; Wright, D. L. Chem. Soc. Rev. 2006, 35, 605-621.
CrossRef - Moeller, K. D. Tetrahedron 2000, 56, 9527-9554.
CrossRef - Francke, R.; Little, R. D. Chem. Soc. Rev. 2014, 43, 2492-2521.
CrossRef - Steckhan, E. Organic syntheses with electrochemically regenerable redox systems, Electrochemistry I; Springer Berlin Heidelberg: Berlin: Heidelberg, 1987.
CrossRef - Suga, S.; Watanabe, M.; Yoshida, J.-I. J. Am. Chem. Soc. 2002, 124, 14824-14825.
CrossRef - Suga, S.; Watanabe, M.; Song, C.-H.; Yoshida, J.-I. Electrochem. 2006, 74, 672-679.
CrossRef - Little, R. D.; Fox, D. P.; Van Hijfte, L.; Dannecker, R.; Sowell, G.; Wolin, R. L.; Moens, L.; Baizer, M. M. J. Org. Chem. 1988, 53, 2287-2294.
CrossRef - Becking, L.; Schäfer, H. J. Tetrahedron Lett. 1988, 29, 2797-2800.
CrossRef - Bakkali, H.; Marie, C.; Ly, A.; Thobie‐Gautier, C.; Graton, J.; Pipelier, M., Sengmany, S.; Léonel, E.; Nédélec, J. Y.; Evain, M. Functionalized 2, 5‐Dipyridinylpyrroles by Electrochemical Reduction of 3, 6‐Dipyridinylpyridazine Precursors; Wiley Online Library: 2008.
CrossRef - Tanaka, H.; Tokumaru, Y.; Fukui, K.-i.; Kuroboshi, M.; Torii, S.; Jutand, A.; Amatore, C. Synth. 2009, 2009, 3449-3459.
CrossRef - Kawamata, Y.; Yan, M.; Liu, Z.; Bao, D.-H.; Chen, J.; Starr, J. T.; Baran, P. S. J. Am. Chem. Soc. 2017, 139, 7448-7451.
CrossRef - Liu, K.; Tang, S.; Huang, P.; Lei, A. Nature Commun. 2017, 8, 775.
CrossRef - Qiu, Y.; Struwe, J.; Meyer, T. H.; Oliveira, J. C.; Ackermann, L. Chem. Eur. J. 2018, 24, 12784-12789.
CrossRef - Röckl, J. L.; Schollmeyer, D.; Franke, R.; Waldvogel, S. R. Angew. Chem. Int. Ed. 2020, 59, 315-319.
CrossRef - Volke, J.; Liška, F. Electrochemistry in Organic Synthesis; Springer Berlin Heidelberg: Berlin, Heidelberg, 1994.
CrossRef - Bard, A. J.; Faulkner, L. R.; White, H. S., Electrochemical methods: fundamentals and applications; Wiley: 2022.
- Bard, A.; Faulkner, L., Electrochemical methods: fundamentals and applications Wiley: New York 2001.
- Kirste, A.; Elsler, B.; Schnakenburg, G.; Waldvogel, S. R. J. Am. Chem. Soc. 2012, 134, 3571-3576.
CrossRef - Morofuji, T.; Shimizu, A.; Yoshida, J.-I. J. Am. Chem. Soc. 2013, 135, 5000-5003.
CrossRef - Xiao, H.-L.; Yang, C.-W.; Zhang, N.-T.; Zeng, C.-C.; Hu, L.-M.; Tian, H.-Y.; Little, R. D. Tetrahedron 2013, 69, 658-663.
CrossRef - Rosen, B. R.; Werner, E. W.; O’Brien, A. G.; Baran, P. S. J. Am. Chem. Soc. 2014, 136, 5571-5574.
CrossRef - Röckl, J. L.; Schollmeyer, D.; Franke, R.; Waldvogel, S. R. Angew. Chem. Int. Ed. 2020, 59, 315-319.
CrossRef - Kang, J.-C.; Tu, Y.-Q.; Dong, J.-W.; Chen, C.; Zhou, J.; Ding, T.-M.; Zai, J.-T.; Chen, Z.-M.; Zhang, S.-Y. Org. Lett. 2019, 21, 2536-2540.
CrossRef - Steckhan, E. Organic syntheses with electrochemically regenerable redox systems. in electrochemistry I; Springer: 2005.
- Nutting, J. E.; Rafiee, M.; Stahl, S. S. Chem. Rev. 2018, 118, 4834-4885.
CrossRef - Wang, F.; Stahl, S. S. Acc. Chem. Res. 2020, 53, 561-574.
CrossRef - Rafiee, M.; Konz, Z. M.; Graaf, M. D.; Koolman, H. F.; Stahl, S. S. ACS Catal. 2018, 8, 6738-6744.
CrossRef - Rafiee, M.; Miles, K. C.; Stahl, S. S. J. Am. Chem. Soc. 2015, 137, 14751-14757.
CrossRef - Goes, S. L.; Mayer, M. N.; Nutting, J. E.; Hoober-Burkhardt, L. E.; Stahl, S. S. J. Chem. Educ. 2021, 98, 600-606.
CrossRef - Qian, X.-Y.; Li, S.-Q.; Song, J.; Xu, H.-C. ACS Catal. 2017, 7, 2730-2734.
CrossRef - Siu, J. C.; Sauer, G. S.; Saha, A.; Macey, R. L.; Fu, N.; Chauviré, T.; Lancaster, K. M.; Lin, S. J. Am. Chem. Soc. 2018, 140, 12511-12520.
CrossRef - Wang, Z.-H.; Gao, P.-S.; Wang, X.; Gao, J.-Q.; Xu, X.-T.; He, Z.; Ma, C.; Mei, T.-S. J. Am. Chem. Soc. 2021, 143, 15599-15605.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


