A Conceptual DFT Based Analysis of Thermodynamic and Global Reactivity Parameter Indices for Pyrogallol Based Complex with Al3+, Cr3+ and Fe3+ Metal ions
Pramod Kumar1 , Amardeep1, Meenakshi1, Vijay Dangi1*, Jitender2, and Brahamdutt Arya3,4*
, Amardeep1, Meenakshi1, Vijay Dangi1*, Jitender2, and Brahamdutt Arya3,4*
1Department of Chemistry, Baba Mastnath University, Rohtak, Haryana, India.
2Department of Chemistry, R.P.S. Degree College, Mahendergarh, Haryana, India.
3Department of Higher Education, Shiksha Sadan, Sec – 5, Panchkula, Haryana, India.
4Y and Y Nanotech Solutions Private Limited, Rohtak, Haryana, India.
Corresponding Author E-mail: brahm.chem@gmail.com and 91dangi@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/400214
Article Received on : 21 Mar 2024
Article Accepted on :
Article Published : 22 Apr 2024
Reviewed by: Dr. sathyanarayanamoorthi venkatachalam
Second Review by: Dr. Hakim MADANI
Final Approval by: Dr. Tanay Pramanik
Heavy metal ions are a major concern due to their ability to harm both people and the environment. Heavy metal ion toxicity has been shown to be significantly reduced by schiff base biomimetic ligands. We have investigated the thermodynamic and stability parameters for Schiff base ligand MEP - trivalent metal ions (Al3+, Cr3+, and Fe3+) complexes based on pyrogallol using DFT and TD-DFT methodologies. In order to propose the function of these metal-ligand complexes in various biological, sensing, and catalytic applications, we have also conducted conceptual density functional theory analysis. We have given the capabilities of MEP-Al3+, MEP-Cr3+, and MEP-Fe3+ complexes to a wide range of industrial and research-based applications, with the primary motto of "Waste to riches" as our guiding principle. TD-DFT, conceptual DFT, and DFT were used in our joint investigation, which led to this conclusion.
KEYWORDS:Conceptual DFT; DFT; Heavy Metal; Pyrogallol; Remediation; Schiff base; Toxicity
Download this article as:| Copy the following to cite this article: Kumar P, Amardeep, Meenakshi, Vijay Dangi, Jitender, Brahamdutt Arya. A Conceptual DFT Based Analysis of Thermodynamic and Global Reactivity Parameter Indices for Pyrogallol Based Complex with Al3+, Cr3+ and Fe3+ Metal ions. Orient J Chem 2024;40(2). |
| Copy the following to cite this URL: Kumar P, Amardeep, Meenakshi, Vijay Dangi, Jitender, Brahamdutt Arya. A Conceptual DFT Based Analysis of Thermodynamic and Global Reactivity Parameter Indices for Pyrogallol Based Complex with Al3+, Cr3+ and Fe3+ Metal ions. Orient J Chem 2024;40(2). Available from: https://bit.ly/3JOfxzv |
Introduction
Aluminium, chromium, and iron are examples of heavy metals that are important environmental pollutants1. These metals come from industrial processes like mining, smelting, and manufacturing, which discharge them into the environment and cause them to build up in soil, water, and the atmosphere2. In many industrial uses, iron, aluminium, and chromium are essential elements that are found in many parts of the environment. However, when present in excessive levels or in certain hazardous forms, their toxicity poses concerns to ecological stability and human health1,3.
Iron is a crucial element in the process of blood production. However, too much iron can be dangerous since too much iron can cause hemochromatosis, also called iron overload condition4. Moreover, iron buildup in organs and tissues can result in a host of grave health complications, including diabetes, heart troubles, and liver illness5. Furthermore, elevated iron concentrations are one of the main causes of several environmental problems, such as eutrophication, which has a gravely negative impact on aquatic life by degrading water quality6.
Similarly, exposure to high concentrations of aluminium has been linked to a number of fatal illnesses, including Alzheimer’s and some forms of cancer, as well as inducing a particular neurological toxicity in humans7,8. Furthermore, increased aluminium concentrations have the potential to seriously harm the environment by influencing the water and soil. According to current research, aluminium poisoning can have a detrimental impact on forest ecosystems and aquatic life by altering pH levels and having toxic effects on fish and plant roots9. However, chromium can exist in a variety of oxidation states and is a major source of industrial pollutants. Trivalent chromium (Cr) is a crucial nutrient, but hexavalent Cr is exceedingly dangerous and carcinogenic10. Direct contact with hexavalent chromium is connected not only to the development of malignancies of the respiratory system but also to skin irritations and ulcers11. Because chromium is hazardous, especially in its hexavalent form, both the water’s quality and the species that inhabits it are at risk. It is a component that negatively impacts the biological and ecological species that reside in ecosystems and adds to their degradation12.
It is difficult, but necessary, to reduce the toxicity of heavy metals like iron, chromium, and aluminium in the context of environmental and public health management. Despite being naturally occurring and essential to many biological processes, these metals can be harmful to the environment and human health when present in excessive concentrations as pollutants.
Many research groups have been working hard over the past few years to reduce the contamination caused by these heavy metal ions. Many scientists are interested in the use of organic Schiff base ligands for the efficient detection and capture of heavy metal ions13,14. The primary benefit of employing Schiff base ligand for the mitigation of heavy metal pollution is that the metal-ligand complex that remains after the heavy metal ions are removed can be utilized for a range of purposes, including biological activity, drug delivery, optical device fabrication, catalysis, and the development of novel pharmaceutically active molecules13.
In a recent work, we have detailed the pH-dependent experimental complex forming capabilities of Schiff base ligand MPC for the efficient and successful detection and complexation of trivalent metal ions15. We intended to use these trivalent metal-ligand complexes in catalysis, sensing, and other applications as a continuation of our earlier work. With the motto “waste to wealth,” our goal is to create innovative organic biomimetic systems that mitigate the harmful effects of heavy metal ions while also using them as outstanding materials for catalysis and sensing applications.
In the current work, we have used DFT and TD-DFT analysis to examine the thermodynamic and stability investigations for MEP-Al3+, MEP-Cr3+, and MEP-Fe3+ complexes. Furthermore, we have used conceptual density functional theory to critically analyze the global reactivity parameter indices in order to access the potential for employment of these metal-ligand complexes in specific applications. Additionally, we have demonstrated the molecule electrostatic surface potential and analyze the preferred sites of electrophilic and nucleophilic attacks during the organic transition. We may suggest that MEP-Al3+, MEP-Cr3+, and MEP-Fe3+ complexes are involved in a variety of catalysis, sensing, and other biological applications based on our collective analysis of DFT, TD-DFT, and conceptual DFT investigations.
Molecular Modelling
Using Gaussian 09 software16, all calculations were carried out on an 11th generation Intel(R) Core (TM) i7-11700K @ 3.40GHz machine. The density functional theory (DFT) has been utilized in this study to optimize the suggested compounds. Furthermore, we have used B3LYP hybrid parameter with computational calculations to access the different structural properties of metal complexes in the gas phase17. Following the optimization of the molecules’ geometry, vibrational frequency calculations were performed at the same theoretical level to verify that the structures are actual minima, that is, with minimum energies.
Theoretical aspects
We have analyzed a number of factors related to the molecules’ overall chemical reactivity in the current study. A variety of reactivity metrics, such as chemical potential, ionization potential, electron affinity, chemical hardness, chemical softness, electrophilic index, nucleophilic index, and so on, can generally be used to express the chemical reactivity of a molecule or complex18. The Frontier Molecular Orbital (FMO) theory can be used to investigate the computed values of these parameters19.
In summary, the FMO theory postulates that a molecule’s interactions with other molecules and the potential for related structural or functional changes happen during a reaction and are dependent on the electron density of the molecular orbitals. Stated differently, we can use FMO theory to estimate the excitation properties of the molecule and then compute the quantum parameters to obtain the total chemical reactivity, which is commonly referred to as the global reactivity indices18,20. Additionally, it can be assumed that a molecule will be more reactive to sensitive transformations if there is a smaller energy difference between the HOMO and LUMO orbitals. Conversely, a wide energy difference between HOMO and LUMO indicates that the molecule is stable and could be associated with little reactivity or no involvement in chemical reactions. When taken as a whole, the global reactivity indices can provide insight into the structural properties, chemical reactivity, and potential bonding sites that exist throughout the reaction. Global reactivity measures often serve as a gauge for a molecule’s overall chemical behaviour.
The energy needed to extract an electron from the complex highest occupied molecular orbital is known as the ionization potential (IP)21. The complex’s capacity to engage in redox processes and contribute electrons is largely predicted by this characteristic. Moreover, Koopman’s approximation (as demonstrated in eq. 1), which stipulates that ionization potential is negative of the energy of highest occupied molecular orbital (EHOMO)22, provides a straightforward expression for ionization potential. Conversely, electron affinity (EA) quantifies the degree to which a molecule or complex manages to hold onto an extra electron21. Koopman’s estimate, which asserts that electron affinity is the negative of the energy of the lowest unoccupied molecular orbital (ELUMO), can also be used mathematically to calculate EA (as shown in eq. 2).
Furthermore, as described by Pauling and Mulliken, the chemical potential, or μ, measures the tendency of the electron cloud to escape from the molecule and is equal to the opposite of the electronegativity, or χ (tendency to attract a shared pair of electrons)23–25. To compute the electronegativity (χ) and chemical potential (μ), use a straightforward approximation between IP and EA, which can be stated in equations 3 and 4, respectively.
Additionally, as demonstrated by eq. 5, we have used the function given by Parr and Pearson to determine the hardness (η) of the molecule/complex23. In a nutshell, chemical hardness is a measurement of how resistant a metal-ligand complex is to changes in electron density brought about by molecular interactions or chemical reactions. In general, it can be obtained from the first derivative of the chemical potential, where N is limited by a set external potential. Conversely, softness (S) can be defined as the reciprocal of hardness (η) and is a quantitative indicator of the amount of electron cloud that has diffused from the molecule24. It can be stated mathematically as eq. 5 correspondingly.

where E(HOMO)and E(LUMO)are the energies of the HOMO and LUMO orbitals respectively.
Furthermore, both softness (S) and hardness (η) can be represented in terms of electronic affinity (EA) and ionization potentials (IP) in accordance with Pearson’s theory of acids and bases26. Equation 6 displays the mathematical expressions correspondingly.
The electrophile index (ω), in addition to chemical hardness and softness, is a significant characteristic that may be assessed using density functional theory. The ability of a molecule to function as an electrophile, or electron acceptor, in chemical reactions is indicated by the electrophile index. The electrophilicity index has been developed by Parr and colleagues as a function of hardness and chemical potential. The electrophile index (ω) can be calculated, in general, using the mathematical method found in equations 7 respectively20,27. The nucleophilicity index (NI), on the other hand, quantifies a molecule’s or complex’s propensity to give two electrons in order to create a new chemical bond. In mathematics, NI is the ionization potential’s negative value, and it is typically stated as indicated in eq. 8 accordingly20.
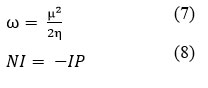
Results and Discussion
Theoretical Studies of Ligand and Their Metal Complexes
To investigate the interactions and complexation behavior of the ligand MEP with metal ions, theoretical research was done on the metal complexes of the ligand MEP. First, the molecular mechanics (MM) methodology was used to optimize the complex’s initial geometry. This was followed by a re-optimization using a semi-empirical method using PM6 parameter28. Furthermore, the optimized structures were once again optimized utilizing the Density Functional Theory (DFT) approach in order to achieve the final geometries of the complexes with least strain energy structures29. Frequency computations were used to analyse the complexes; the lack of imaginary frequencies confirmed the energy minima for the geometry and showed that the molecular geometry was fully optimized. Only the complexes in the gas phase were used in all of the computations. The optimized structures for the complexes including trivalent metal ion complexes Al3+, Cr3+, and Fe3+, respectively, with ligand MEP are shown in Figure 1. Additionally, using DFT, the energies of the optimized structures for the ligand MEP and the trivalent metal ion complexes Al3+, Cr3+, and Fe3+ were determined to be -1875.99 a.u., -2677.60 a.u., and – 2896.76 a.u., respectively.
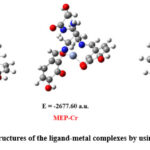 |
Figure 1: Optimized structures of the ligand-metal complexes by using Semi-empirical/PM6 |
DFT was used to calculate a range of thermodynamic properties, the energy of frontier MO and band gaps, and other important reactivity factors at different levels.
Thermodynamic Parameters and Relative Stability
Gibbs free energy (G), entropy (S), and enthalpy (H) are three thermodynamic parameters that are crucial to understanding the overall stability of any metal ligand complex. For the ligand MEP with trivalent metal ions Al3+, Cr3+, and Fe3+ complexes, we computed the thermodynamic parameters (enthalpy, entropy, and gibbs free energy) using the DFT approach in the current study. The results are tabulated in Table 1 and graphically displayed in Figure 2.
Table 1: Thermodynamic parameters (Enthalpy, Gibbs free energy and Entropy) of MEP metal complexes.
|
Complexes |
H (a.u.) |
G (a.u.) |
S (cal/mol) |
|
MEP-Al+3 |
-1875.56 |
-1875.65 |
196.88 |
|
MEP-Cr+3 |
-2677.17 |
-2677.26 |
195.48 |
|
MEP-Fe+3 |
-2896.33 |
-2896.42 |
195.83 |
The order of stability of the complexes is indicated by all the characteristics, which are determined in the gaseous phase. The findings show that the MEP-Fe3+ combination has the lowest enthalpy value (-2896.33 a.u.), followed by the MEP-Cr3+ complex (-2677.17 a.u.) and the MEP-Al3+ complex (-1875.56 a.u.) complexes, in that order. Furthermore, MEP-Fe3+ (-2896.42 a.u.) < MEP-Cr3+ (-2677.26 a.u.) < MEP-Al3+ (-1875.65 a.u.) is the trend that the gibbs free energy follows. These data sets unambiguously show that, among the three metal ions under consideration, the ligand MEP is more stable to the complex Fe3+ metal ion. Figure 2 displays a comparative graphical depiction of thermodynamic parameters.
 |
Figure 2: A comparative graphical analysis of major thermodynamic parameters viz. enthalpy (H), entropy (S) and gibbs free energy (G) calculated by using density functional theory with B3LYP parameter for of MEP- Al3+, MEP-Cr3+ and MEP-Fe3+metal complexes respectively. |
Frontier Molecular Orbitals (FMO) Calculation
We have used TD-DFT calculations utilizing the B3LYP hybrid functional to determine the energies of frontier molecular orbitals, or the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), for each of the metal complexes, namely MEP- Al3+, Cr3+, and Fe3+. Additionally, using the corresponding energies of HOMO and LUMO, the relative energy gap between them was computed for each of the three metal complexes.
Table 2: Energies of HOMO and LUMO molecular orbitals and the energy gaps [ΔE (eV)] of MEP metal complexes.
|
Complexes |
EHOMO (eV) |
ELUMO (eV) |
ΔE (eV) |
|
MEP-Al+3 |
-2.75 |
1.47 |
4.22 |
|
MEP-Cr+3 |
-5.66 |
-3.35 |
2.31 |
|
MEP-Fe+3 |
-5.58 |
-3.54 |
2.04 |
Table 2 displays the computed HOMO and LUMO energies together with the corresponding ΔE values (relative energy gap between HOMO and LUMO) for the Al3+, Cr3+, and Fe3+ complexes. Moreover, Fig. 3 shows a comparative visual analysis of the associated HOMO and LUMO with their corresponding band gap for the Al3+, Cr3+, and Fe3+ complexes. The MEP-Fe3+ complex was found to have the lowest energy gap (ΔE = 2.04 eV), indicating that among the three complexes, it is the most reactive. However, among the three metal-ligand complexes under investigation, the MEP-Al3+ combination has the largest value of ΔE (4.22 eV), indicating its least reactive behavior.
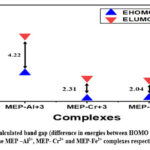 |
Figure 3: Calculated band gap (difference in energies between HOMO and LUMO) of the MEP –Al3+, MEP- Cr3+ and MEP-Fe3+ complexes respectively. |
Band gaps in organic semiconductor materials typically lie between 1 and 5 eV. All three of the metal-ligand complexes in our study fall within the same range and have potential uses in a variety of semi-conductivity-based applications30.
 |
Figure 4: Frontier Molecular Orbitals of the ligand-metal complexes by using DFT/B3LYP/LANL2DZ |
Using DFT-based B3LYP/LANL2DZ functionals, we have further investigated the relative electron density distribution for all trivalent metal-ligand MEP complexes. At a given atomic center in the molecule, the relative electron energy distribution can provide information on the potential location of the HOMO and LUMO electron densities. The HOMO and LUMO electron density distributions for every trivalent metal complex are shown in Figure 4. With close examination of the structures in Figure 4, one can deduce that the HOMO and LUMO electron densities are primarily found on the central metal atom and binding unit, or pyrogallol moiety, of the complexes. Generally speaking, we may predict the electrophilic or nucleophilic behaviour of a particular site by looking at the location of the HOMO and LUMO electron densities. The majority of the time, a site’s ability to operate as a nucleophile and the ease with which electrons can participate in the reaction are linked to the localization of the HOMO electron density. However, the presence of a suitable electrophilic site can be linked to the position of the LUMO electron density31.
Global Indices of Reactivity
We investigated several different properties in the present study that are related to the molecules’ overall chemical reactivity. A combination of factors, including chemical potential, ionization potential, electron affinity, chemical hardness, chemical softness, electrophilic index, and nucleophilic index, among others, can generally be used to indicate the chemical reactivity of a molecule or complex20. The work of computing the computed values of these parameters can be completed by applying the Frontier Molecular Orbital (FMO) theory. In summary, we utilized the DFT and TD-DFT computations to derive distinct data points, and subsequently computed multiple global reactivity indices, including IP, EA, μ, χ, η, S, ω, and N.I., utilizing equations 1 through 8.
In spite of the fact that the wave function-based molecular orbital technique typically yields very accurate findings, its usefulness is limited due to the fact that related correlation effects interfere with it and the demand for an excessively complicated computational setup32. An additional avenue for systematically investigating the reactivity parameters is provided by density functional theory. All ground state information is computed using electron density as the foundation of operation17. Furthermore, since the reactive parameters were determined as a function of both total energy and electron density, the reliance on the spatial and spin coordinates of each and every electron in the molecules is no longer an issue. Parr and Yang originally reported on the interpretation of chemical reactivity using this electron density-based method, which is commonly referred to as “conceptual density functional theory”27. Table 3 presents a comparative analysis of all these reactive characteristics for Al3+, Cr3+, and Fe3+ complexes.
Table 3: Ionization potential (IP, eV), electron affinity (EA, eV), chemical potential (μ, eV), chemical hardness (η, eV), softness (S, eV-1) and overall electrophile index (ω, eV) of metal complexes.
|
Complexes |
IP (eV) |
EA (eV) |
μ(eV) |
χ(eV) |
η(eV) |
S(eV) |
ω(eV) |
N.I. |
|
MEP-Al+3 |
2.75 |
-1.47 |
-0.64 |
0.64 |
2.11 |
0.47 |
0.097 |
-2.75 |
|
MEP-Cr+3 |
5.66 |
3.35 |
-4.51 |
4.51 |
1.16 |
0.86 |
8.77 |
-5.66 |
|
MEP-Fe+3 |
5.58 |
3.54 |
-4.56 |
4.56 |
1.02 |
0.98 |
10.19 |
-5.58 |
For the purpose of determining the total chemical reactivity, it is important to conduct a comprehensive analysis of all potential reactivity parameters. It is necessary to investigate the ionisation potential (IP) and electron affinity (EA) of a complex in order for it to exhibit the capacity to attract holes and electrons, respectively. Lower IP values often indicate easier electron removal (or hole injection), and on the other hand, larger EA values indicate easier electron acceptance. The IP value for the MEP-Al3+ complex in Table 3 is 2.75 eV, lowest among the three metal-ligand complex values. This suggests that the MEP-Al3+ complex has a stronger tendency to exhibit electron removal than the other two metal-ligand complexes. However, out of all the metal ligand complexes, the MEP-Cr3+ combination has the highest IP value of 5.66 eV, indicating that it has the least easy electron removal behaviour. To put it briefly, the three metal-ligand complexes are arranged as follows: MEP-Al3+ < MEP-Fe3+ < MEP-Cr3+ in increasing order of IP.
The graphical representation of the electron affinity and IP value for the Al3+, Cr3+, and Fe3+ complexes, respectively, is shown in Figure 5. IP observations indicate that EA will follow the opposite pattern. The sequence of EA was discovered to be MEP-Al3+ < MEP-Cr3+ < MEP-Fe3+, in that order. There is a correlation between diminishing electronegativity and a decrease in the tendency to donate electrons.
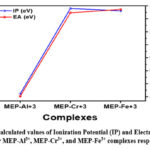 |
Figure 5: Calculated values of Ionization Potential (IP) and Electron Affinity (EA) for MEP-Al3+, MEP-Cr3+, and MEP-Fe3+ complexes respectively. |
Furthermore, electronegativity and chemical potential are two of the most important reactivity descriptors that are used in order to gain access to the potential interactions that the metal ligand complex may have with other reactive species21. Electronegativity, in a broad sense, refers to the tendency of a complex to attract a pair of electrons that are previously shared with it. To the contrary, the capacity of the electron cloud to escape may be related to chemical potential in some way33. After closely examining the value of electronegativity (EN) in table 3, we discovered that MEP-Fe3+ has the highest value, 4.56 eV, indicating that it is more electrophilic than the other metal-ligand complexes that are being studied. However, out of all the metal-ligand complexes, MEP-Al3+ had the lowest EN value (0.64 eV), indicating a less electrophilic nature. EN is arranged as follows: MEP-Al3+ < MEP-Cr3+ < MEP-Fe3+. On the other hand, the chemical potential follows the reverse pattern, with MEP-Fe3+ being preceded by MEP-Cr3+ and then by MEP-Al3+. In comparison to the other three metal-ligand complexes that are currently under investigation, MEP-Al3+ has the potential to demonstrate the highest degree of escape the electron cloud. The computed values of electronegativity and chemical potential were shown in Figure 6, respectively.
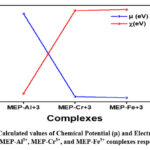 |
Figure 6: Calculated values of Chemical Potential (μ) and Electronegativity (χ) for MEP-Al3+, MEP-Cr3+, and MEP-Fe3+ complexes respectively. |
In addition, it is interesting to note that the higher the electronegativity of a complex, the greater the likelihood that it would exhibit an electrophilic feature. To phrase it another way, the electrophilicity index (EI) will have a larger value when the electronegativity is higher34. Consequently, the electrophilic index will exhibit the same pattern as EN, specifically, MEP-Al3+ < MEP-Cr3+ < MEP-Fe3+. With an EI value of 10.19, the MEP-Fe3+ complex is the most electrophilic of all; in contrast, the MEP-Al3+ complex has the lowest electrophilicity, with an EI value of 0.097. The electrophilic and nucleophilic indices of the MEP-Al3+, MEP-Cr3+, and MEP-Fe3+ complexes are shown in Figure 7, respectively.
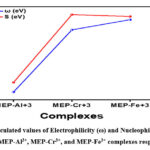 |
Figure 7: Calculated values of Electrophilicity (ω) and Nucleophilicity Index (N.I.) for MEP-Al3+, MEP-Cr3+, and MEP-Fe3+ complexes respectively. |
Nucleophilicity index (NI), on the other hand, follows the trends, which are MEP-Cr3+ < MEP-Fe3+ < MEP-Al3+. It’s interesting to note that the MEP-Al3+ complex has the highest NI value of all, -2.75, indicating a larger degree of nucleophilic behaviour in the reactions. However, out of all the metal-ligand complexes being studied, MEP-Cr3+ has the lowest nucleophilic value, or -5.66, indicating the least nucleophilic behaviour.
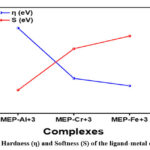 |
Figure 8: Hardness (η) and Softness (S) of the ligand-metal complexes |
We have used the idea defined by Parr and Pearson to access the hardness (η) and softness (S)24. This means that the change in chemical potential with N while an external potential is fixed can be used to characterise the hardness of the complex18. After closely examining table 3, we have discovered that, out of the three metal-ligand complexes under investigation, MEP-Al3+ has the greatest hardness value (2.11), suggesting that it is more stable in chemical reactions. However, out of the three metal-ligand complexes under investigation, the MEP-Fe3+ combination has the lowest hardness value (1.02), indicating that it is more reactive.
Generally speaking, there is a relationship between a compound’s hardness and softness and the energy difference between its HOMO and LUMO states, or band gap. Larger energy gaps have often been observed to increase a compound’s resistance to the charge transfer process because they increase stability, hardness, and reduce reactivity. According to our research, the metal-ligand complexes’ energy gap, hardness, and stability generally follow the following order: MEP-Fe3+ < MEP-Cr3+ < MEP-Al3+. Conversely, the order MEP-Al3+ < MEP-Cr3+ < MEP-Fe3+ represents the common tendency for the reactivity and softness of the metal-ligand complexes. The hardness and softness sequence that the metal-ligand complexes in our current study followed is shown in Figure 8.
Determination of Molecular Electrostatic Potential surface (MEPs)
In this work, we have used the DFT based B3LYP optimized geometry functionals to analyze the molecular electrostatic potential (MEP) surface for MEP-Al3+, MEP-Cr3+, and MEP-Fe3+ complexes. The molecular electrostatic potential (MEP) surfaces for each of the three metal-ligand complexes are shown in Figure 9. The sections of the system that are particularly rich in electrons and those that are deficient in electrons are identified using the molecular electrostatic potential surface, which is useful in recognizing the region of electrophilic and nucleophilic assault35. Red indicates a zone of high electron density on the molecular electrostatic potential (MEP) surface. The color blue is linked to a zone of high electron density, which is linked to a positive electrostatic potential, whereas this zone is related with a negative electrostatic potential36. The neutral area is represented by the color green, which indicates zero electrostatic potential. The presence of the green zone on every atom in the aluminium complex is proof that the molecule’s electrostatic potential is equal to zero.
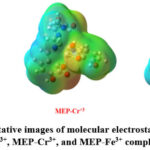 |
Figure 9: Representative images of molecular electrostatic potential (MEP) surfaces MEP-Al3+, MEP-Cr3+, and MEP-Fe3+ complexes respectively. |
In contrast, the red color of the oxygen atoms in the chromium complex denotes a high electron density for oxygen atoms and a low electron density for hydrogen and carbon atoms, respectively. In addition, the iron complex has red sections for the oxygen atoms, blue regions for the nitrogen atoms, and green regions for the carbon and hydrogen atoms. The iron complex encompasses all of these areas. In all of the complexes under investigation, heteroatoms like oxygen and nitrogen atoms have a substantial amount of electron-rich density, whereas carbon and hydrogen atoms have electron-deficient sites.
Conclusions
The thermodynamic and chemical reactivity parameters of the MEP-Al3+, MEP-Cr3+, and MEP-Fe3+ complexes were investigated in the current study using DFT, TD-DFT, and conceptual DFT based analysis, respectively. The MEP-Fe3+ complex is more stable than the Al3+ and Cr3+ complexes, respectively, based on DFT analysis for the computation of the energy, thermodynamic, and electronic properties. Furthermore, the MEP-Fe+3 complex showed a low value of Gibbs free energy, or G = -2896.42, suggesting that it was more thermodynamically stable. Furthermore, among all the metal-ligand complexes under consideration, the MEP-Fe3+ complex seems to be the softest and reactive based on conceptual DFT analysis. Based on the current investigation, we have shown how to analyse metal ligand complexes globally using chemical reactive descriptors for their possible use in a variety of applications, including chelation therapy, semiconductor devices, and catalysis.
Acknowledgement
Dr. Minati Baral, professor at NIT Kurukshetra, India, are suitably acknowledged for their unwavering support and direction.
Conflict of Interest
Regarding financial and relationships to organisations, none of the
authors have any conflicts of interest.
References
- Abd Elnabi, M. K.; Elkaliny, N. E.; Elyazied, M. M.; Azab, S. H.; Elkhalifa, S. A.; Elmasry, S.; Mouhamed, M. S.; Shalamesh, E. M.; Alhorieny, N. A.; Abd Elaty, A. E.; Elgendy, I. M.; Etman, A. E.; Saad, K. E.; Tsigkou, K.; Ali, S. S.; Kornaros, M.; Mahmoud, Y. A.-G. Toxics 2023, 11 (7), 580.
CrossRef - Balali-Mood, M.; Naseri, K.; Tahergorabi, Z.; Khazdair, M. R.; Sadeghi, M. Front. Pharmacol. 2021, 12, 643972.
CrossRef - Arao, T.; Ishikawa, S.; Murakami, M.; Abe, K.; Maejima, Y.; Makino, T. Paddy Water Environ. 2010, 8 (3), 247–257.
CrossRef - Eaton, J. W.; Qian, M. Free Radic. Biol. Med. 2002, 32 (9), 833–840.
CrossRef - Papanikolaou, G.; Pantopoulos, K. Toxicol. Appl. Pharmacol. 2005, 202 (2), 199–211.
CrossRef - Saaltink, R. M.; Dekker, S. C.; Eppinga, M. B.; Griffioen, J.; Wassen, M. J. Plant Soil 2017, 416 (1–2), 83–96.
CrossRef - Exley, C. Morphologie 2016, 100 (329), 51–55.
CrossRef - Closset, M.; Cailliau, K.; Slaby, S.; Marin, M. Int. J. Mol. Sci. 2021, 23 (1), 31.
CrossRef - Gensemer, R. W.; Playle, R. C. Crit. Rev. Environ. Sci. Technol. 1999, 29 (4), 315–450.
CrossRef - Costa, M.; Klein, C. B. Crit. Rev. Toxicol. 2006, 36 (2), 155–163.
CrossRef - Shelnutt, S. R.; Goad, P.; Belsito, D. V. Crit. Rev. Toxicol. 2007, 37 (5), 375–387.
CrossRef - Jaishankar, M.; Tseten, T.; Anbalagan, N.; Mathew, B. B.; Beeregowda, K. N. Interdiscip. Toxicol. 2014, 7 (2), 60–72.
CrossRef - More, M. S.; Joshi, P. G.; Mishra, Y. K.; Khanna, P. K. Mater. Today Chem. 2019, 14, 100195.
CrossRef - Abdel-Rahman, L. H.; Basha, M. T.; Al-Farhan, B. S.; Alharbi, W.; Shehata, M. R.; Al Zamil, N. O.; Abou El-ezz, D. Molecules 2023, 28 (12), 4777.
CrossRef - Amardeep, A.; Dangi, V.; Kumar, P.; Meenakshi, M.; Baral, M.; Arya, B.; Sheoran, T. Orient. J. Chem. 2024, 40 (1), 274–280.
CrossRef - Duke, B. J.; O’Leary, B. J. Chem. Educ. 1992, 69 (7), 529.
CrossRef - Xu, Y.-C.; Li, N.; Yan, X.; Zou, H.-X. Environ. Sci. Pollut. Res. 2023, 30 (40), 91780–91793.
CrossRef - Pal, R.; Chattaraj, P. K. J. Indian Chem. Soc. 2021, 98 (1), 100008.
CrossRef - Yu, J.; Su, N. Q.; Yang, W. JACS Au 2022, 2 (6), 1383–1394.
CrossRef - Domingo, L.; Ríos-Gutiérrez, M.; Pérez, P. Molecules 2016, 21 (6), 748.
CrossRef - Zhan, C.-G.; Nichols, J. A.; Dixon, D. A. J. Phys. Chem. A 2003, 107 (20), 4184–4195.
CrossRef - Shankar, R.; Senthilkumar, K.; Kolandaivel, P. Int. J. Quantum Chem. 2009, 109 (4), 764–771.
CrossRef - Gázquez, J. L. In; Sen, K. D., Ed.; Structure and Bonding; Springer-Verlag: Berlin/Heidelberg, 1993; Vol. 80, pp 27–43.
- Xu, H.; Xu, D. C.; Wang, Y. ACS Omega 2017, 2 (10), 7185–7193.
CrossRef - Franco-Pérez, M.; Gázquez, J. L. J. Phys. Chem. A 2019, 123 (46), 10065–10071.
CrossRef - Ayers, P. W.; Mohamed, M.; Heidar‐Zadeh, F. In; Liu, S., Ed.; Wiley, 2022; pp 263–279.
CrossRef - Chakraborty, D.; Chattaraj, P. K. Chem. Sci. 2021, 12 (18), 6264–6279.
CrossRef - Bernhardt, P. V.; Comba, P. Inorg. Chem. 1992, 31 (12), 2638–2644.
CrossRef - Wang, W.; Zhu, J.; Huang, Q.; Zhu, L.; Wang, D.; Li, W.; Yu, W. Molecules 2024, 29 (2), 308.
CrossRef - Aderne, R. E.; Borges, B. G. A. L.; Ávila, H. C.; Von Kieseritzky, F.; Hellberg, J.; Koehler, M.; Cremona, M.; Roman, L. S.; Araujo, C. M.; Rocco, M. L. M.; Marchiori, C. F. N. Mater. Adv. 2022, 3 (3), 1791–1803.
CrossRef - Domingo, L. Molecules 2016, 21 (10), 1319.
CrossRef - Hancock, R. D. Acc. Chem. Res. 1990, 23 (8), 253–257.
CrossRef - Bartlett, R. J.; Grabowski, I.; Hirata, S.; Ivanov, S. J. Chem. Phys. 2005, 122 (3), 034104.
CrossRef - Pérez, P.; Domingo, L. R.; Aizman, A.; Contreras, R. In; Elsevier, 2007; Vol. 19, pp 139–201.
CrossRef - Daolio, A.; Pizzi, A.; Calabrese, M.; Terraneo, G.; Bordignon, S.; Frontera, A.; Resnati, G. Angew. Chem. Int. Ed. 2021, 60 (38), 20723–20727.
CrossRef - Kenouche, S.; Sandoval-Yañez, C.; Martínez-Araya, J. I. Chem. Phys. Lett. 2022, 801, 139708.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


