Novel Type 1 5α- Reductase Inhibitors with Antiproliferative Potential on Lncap Cells
Marisa Cabeza1* , Luis A. Menes1, Evelyn Fuentes1, Iván Bahena2, Yvonne Heuze1
, Luis A. Menes1, Evelyn Fuentes1, Iván Bahena2, Yvonne Heuze1
1Departamento of Biological Systems and Agricultural and Animal Production. Universidad Autónoma Metropolitana-Xochimilco, Calzada del Hueso 1100. Col. Villa Quietud, 04960 Mexico City. Mexico
2Department of Experimental Biology. Universidad Autónoma Metropolitana-Iztapalapa San Rafael Atlixco No. 186, Col. Vicentina, Iztapalapa, 09340, Mexico.
Corresponding Author E-mail: marisa@correo.xoc.uam.mx
DOI : http://dx.doi.org/10.13005/ojc/390301
Article Received on : 20 May 2023
Article Accepted on : 21 Jun 023
Article Published : 27 Jun 2023
Reviewed by: Dr. Oguzhan Ozdemir
Second Review by: Dr. Narendra Batham
Final Approval by: Dr. S.A. Iqbal
This study demonstrated the antiproliferative potential of several dehydroepiandrosterone derivatives 2a-b, 3a-f, and 4a-f in LNCaP cells.
LNCaP cells were cultured in the presence of the vehicle, dihydrotestosterone, or testosterone plus 2a-b, 3a-f, 4a-f, or finasteride. At 24 hours, the 3-(4,5-dimethylthiazol-2-yl)-2,5-di phenyltetrazolium bromide- test was performed to determine cell proliferation. In addition, the kinetic of SRD5A1 in these cells was studied in the presence or absence of different concentrations of 2a.
Testosterone and dihydrotestosterone increased the proliferation of LNCaP compared to the vehicle-treated cells. Furthermore, steroids 2a-b, 3a-f, and 4a-f decreased the number of viable cells compared to testosterone treatment. However, finasteride did not affect viability.
LNCaP cells converted radiolabeled testosterone into dihydrotestosterone. This conversion was inhibited by high concentrations of 2a, while at a pM concentration, the conversion increased slightly, suggesting the presence of allosteric sites in SRD5A1.
In conclusion, the three series of derivatives of dehydroepiandrosterone significantly decreased the number of viable LNCaP cells, therefore, showing therapeutic potential to treat metastatic prostate cancer.
dehydroepiandrosterone derivatives; LNCaP cell line; prostate cancer; type 1, 5α-reductase inhibitors; The antiproliferative effect
Download this article as:| Copy the following to cite this article: Cabeza M, Menes L. A, Fuentes E, Bahena I, Heuze Y. Novel Type 1 5α- Reductase Inhibitors with Antiproliferative Potential on Lncap Cells. Orient J Chem 2023;39(3). |
| Copy the following to cite this URL: Cabeza M, Menes L. A, Fuentes E, Bahena I, Heuze Y. Novel Type 1 5α- Reductase Inhibitors with Antiproliferative Potential on Lncap Cells. Orient J Chem 2023;39(3). Available from: https://bit.ly/3Nrfzyv |
Introduction
While countless research groups and pharmaceutical companies have focused their knowledge and effort on early prostate cancer (PC) detection to prevent its spread, most patients with scattered metastases have a poor prognosis at the end of their diagnosis. Worldwide, out of the 21% of men diagnosed with prostate cancer, 16.5% die from metastasis. (Global Cancer Observatory*)
Previous reports have shown that metastatic bone tumor growth can start from malignant migratory cells produced in the prostate. This new tumor can establish itself in the bones [1-4] and begin synthesizing androgens to continue developing. [5] Adaptive androgen signaling in tumor cells may occur because intratumoral androgen biosynthesis or also to the somatic androgen receptor (AR) mutations are carried out. [6-7] It could also be to the AR gene amplification [8] or changes in nuclear coregulators, which make AR constitutively active. [9-11]
The 5α-reductase enzyme (SRD5A) catalyzes the conversion of testosterone (T) to its most active metabolite, which is dihydrotestosterone (DHT, Fig. 1) in androgen-dependent tissues. [12] DHT binds to its cytoplasmic receptor, which shows greater affinity than T. Then the formed complex translocates to the cell nucleus, which binds to the DNA to encode gene expression [13-14].
 |
Figure 1: Testosterone converts to dihydrotestosterone in the LNCaP cell line because of the enzyme SRD5A1 (EC 1.3.99.5). |
Three SRD5A isoenzymes (1, 2, and 3) have been described [15, 16]. SRD5As of types 1 and 3 are present in the LNCaP metastatic prostate cancer cells. These cells take up 4 to 5 pg of T/mg of tissue when incubated with 1 nM T. In addition, they accumulate up to 15 nM of T in the first hour after treatment. However, after 24 h, the T forms glucuronides, and undetectable levels of T could be determined [16]
In the prostate, SRD5A1 and SRD5A2 have been well characterized. These isoenzymes possess different kinetic properties. [17, 18] SRD5A2 is the predominant isoenzyme in the normal prostate and shows a higher T-binding affinity than SRD5A1. [18] While in prostatic intraepithelial neoplasms, there has greater activity of SRD5A1 instead of SRD5A2. In addition, SRD5A1 is largely overexpressed in cancers that be recurrent castration. [19] However, in benign prostatic hypertrophy, there is a decrease in SRD5A1 activity, while an increase in SRD5A2 levels has been found. [20]
The fact that SRD5A1 activity is increased in castration-recurrent prostrate tumors would explain their ability to survive and grow in an environment with low androgen content. A very low concentration of T would be sufficient to induce cell proliferation and secretion of prostate antigen (PSA). [19, 20]. This evidence allows us to hypothesize that specific inhibitors of SRD5A1 could be used as a beneficial treatment for recurrent PC after castration and metastatic prostate cancer.
Previously, our group reported the synthesis and activity of different derivatives of dehydroepiandrosterone as specific inhibitors of SRD5A1. [20-22] Therefore, this research has focused on demonstrating that the particular inhibitors of SRD5A1 shown in Table 1 decrease the proliferative activity of LNCaP cells. [12, 19, 24]
 |
Table 1: Structure and biological activity of different SRD5A1 inhibitors. |
Materials and Methods
The (1, 2, 6, 7-3H) testosterone ([3H]T) with a specific activity of 95 Ci/mmol and Mibolerone ([3H] MIB) with a specific activity of 70 Ci/mmol were purchased from Perkin Elmer (Boston, MA). Steraloids (Wilton, NH, USA) supplied MIB, T, and 5α-DHT. LNCaP cells were acquired from ATCC (Scientific Senna, CDMX). Finasteride (FIN) was extracted from Proscar (Merck, Sharp, and Dohme); the tablets were crushed, extracted with chloroform, and the solvent was eliminated in a vacuum; the crude product was purified with silica gel column chromatography. The melting point of the isolated finasteride (252-254 °C) was identical to that reported in the literature [Trapani et al., 2002]. Steroids 2a-b, 3a-f, and 4a-f (Table 1) were synthesized. [22, 23] Dr. Eugene Bratoeff bequeathed all the steroidal molecules presented in this paper to our laboratory to determine their ability to decrease the LNCaP cells’ viability.
Determination of the effect of 2a-b, 3a-f and 4a-f on the proliferation of LNCaP cells
LNCaP cells (passage 25-50) were used at 4X104 cells/well density in 96 well container plates. The cells were allowed to adhere to the plaque for 48 hours in a Roswell Park Memorial Institute medium (RPMI-1640, phenol-free). The medium containing a phenol-free 10% fetal bovine serum (Glibco, Mexico) and 2 mM glutamine was supplemented. The plates were incubated at 37 °C in the humidified atmosphere, with 5% CO2 -95% air in a VWR incubator (Symphony 1.4 A, VWR, CDMX).
The monolayers of LNCaP cells were washed with phosphate-buffered saline (PBS). Then we separately added different concentrations (10-9-10-4) of the steroids 2a-b, 3a-f,and 4a-f (dissolved in 1. 5 % propylene glycol) plus 1 μM T in each of the wells. The vehicle (1.5% propylene glycol) containing T (1 μM), DHT (1 nM), or finasteride (25 μM) [20] was added separately in other wells and maintained as feasibility controls. All trials were quadrupled on three independent experiments.
The plate was incubated for 24 hours under the same conditions described above. The proliferation of LNCaP cells was determined by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-di phenyltetrazolium bromide) method. [25, 26] The formazan crystals formed from the reaction with 12 mM MTT were dissolved in 100 μL of DMSO. The resulting absorbance was measured by spectrophotometry at 570 nm. (Biotek™ Epoch™ Microplate Spectrophotometer, Thermo Fisher Scientific, USA). Finasteride was used as an inhibitor of SRD5A2 activity in the trial. The FDA has previously approved this steroid to treat benign prostatic hyperplasia.
Preparation of solubilized microsomes as a source of SRD5A1
LNCaP cells were cultured to confluence, the medium was removed, and the culture was rinsed with buffer A (sodium phosphate 20 mM, pH 6.5, sucrose 0.3 M, and dithiothreitol (DTT) 1 mM). The cell monolayer was scraped, and the cells were homogenized in a buffer volume A 1:10. Subsequently, the tube containing this mixture was centrifuged at 100,000 xg for 60 minutes to obtain the microsomes. All the previous procedures were performed at 0-4°C.
The tube sediment was solved in a volume of buffer B (sodium citrate 10 mM, potassium chloride 100 mM, DTT 1.0 mM, Lubrol PX 0.1%, glycerol 20, and NADPH 100 μM adjusting the pH = 6.6) according to Levy’s method [27] and incubated in ice for 30 minutes. The tube was then centrifuged at 100,000 xg for an hour. A supernatant aliquot was stored to determine proteins by the Bradford method, and the remainder was kept at -80°C until further trials with inhibitor 2a. The microsome solution (RS) was diluted in cold 20 mM phosphate buffer, pH 6.6, containing 1 μM NADPH, to a protein concentration of 60 μg/10 μL.
Determination of the optimal incubation time
Subsequently, we conducted incubations at different times using 2 nM T[3H], 200 μM NADPH [27], and 1 mM DTT at a final volume of 0.5 mL of 20 mM sodium phosphate buffer, pH 7. 5 and a temperature of 37 °C. The reaction began by adding 60 μg of protein RS as a source of SRD5A1 to determine the optimal conversion time from [3H]T to [3H]DHT. All the trials were prepared in advance and repeated on four separate occasions.
The resulting steroids were extracted with an equal volume of 5X dichloromethane. The solvent volume was collected in a single tube and evaporated to dryness. The extract was diluted in a mixture of 100 μL chloroform/methanol=2:1. This mixture was applied on an HPTLC plate (Keisel gel 60 F254 HPTLC plates) [Arellano et al., 2018] to separate T from DHT. Different steroid standards (T and DHT) were placed on separate lanes on both sides of this plate to identify the steroids produced. This plaque was then developed into a chloroform/acetone mixture = 9:1 [23] and dried at room temperature. Radioactivity in HPTLC was analyzed with a Bioscanner (AR-2000 Imaging Scanner, Washington, DC). The DHT standard was identified by spraying 8% phosphomolybdic acid in methanol on the corresponding plate rails. At the same time, the T standard was detected with a UV lamp (254 nm).
Characterization of 2a inhibition on SRD5A1 activity
Samples containing different concentrations of unlabeled T (2, 5, 7, 11, 16, 20 μM), 2 nM T[3 H], 200 μM NADPH [27], and in the presence or absence of 2a (0.1 nM, 1 nM, and 1 μM) in a final volume of 0. 5 mL of 20 mM sodium phosphate buffer, pH 7.5 were prepared. The reaction began by adding 30 μg of RS (the amount of protein that produces the optimal conversion to DHT, data not shown) and incubating the samples at 37 °C for 17 minutes (Figure 6). As a control, a mixture identical to the previous one was prepared without the SR in the tube. All the assays were repeated on four separate occasions in triplicate.
The formed steroids (T or DHT labeled or not) were separated and identified, as explained in section 2.3.
Prostatic cytosol isolation
The Internal Committee for the Care and Use of Laboratory Animals (CICUAL) of the Autonomous Metropolitan University (UAM, CDMX) previously approved all rat procedures according to the NOM-062-ZOO 1999.
The prostates of the gonadectomized rats, 48 h. before the experiment were dissected, separated, and homogenized in a volume of TEMD buffer (Tris-HCl 40 mM, EDTA 3 mM, and sodium molybdate 20 mM, dithiothreitol 0.5 mM, glycerol 10% at pH 8) plus protease inhibitors (PMSF 2 mM, 10 mg/ml antipain, leupeptin 5 mM). [28]. All these procedures were carried out at 4 °C on ice. The homogenates were centrifuged at 140,000 xg for 60 minutes, and an aliquot of the supernatant (cytosol) was aliquoted for protein determination. The cytosol was then stored at -70 °C until use.
Androgen receptor (AR) competition tests
For competitive binding tests, we follow the method reported above. [29] Briefly, samples containing 4 nM of [3H]MIB plus a range of increasing concentrations (10-10-10-4 M) of unbranded MIB or with each of the steroids 4a-f (Table 1) were incubated at 4 °C for 18 h., in the presence of 5 mg of the prostate rat cytosolic protein, in a final volume of TEMD buffer (40 mM Tris-HCl, 3 mM EDTA and 20 mM sodium molybdate, dithiothreitol 0.5 mM, 10% glycerol at pH 8) containing protease inhibitors 0.5 mL (per triplicate). Then, the [3H]MIB-AR bound complex was obtained using hydroxylapatite (HAP) previously described by Liao et al. [30]. Briefly, we added 500 μL of the HAP suspension to the sample and held it for 15 minutes at 4ºC. After centrifugation at 2500 g, the sediment was washed three times with a 50 nM tris buffer at pH 7.2 to remove the unbound [3H]MIB. The bound [3H]MIB was extracted from the sediment with a volume of ethanol for one hour at room temperature. A 500 μl aliquot was removed from the sample supernatant with the [3H]MIB-AR complex and sent for radioactive counting on a Packard Tri-Carb scintillation counter. Each compound inhibitory concentration 50 (IC50) was calculated from concentration plots versus the binding percentage. The following equation calculated the relative binding affinity:
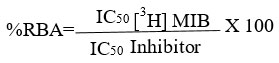
Statistical analysis
The statistical analysis was accomplished using ANOVA One Way and Dunnett’s test to compare the means with JMP version 8 software. P<0.05 value was accounted as a significant difference.
Results and Discussion
Effect of T and DHT on LNCaP cell proliferation
Figure 2 shows the number of viable LNCaP cells in different concentrations of T or DHT after 24 h. of treatment. The results indicated that a concentration of 1 μM T increased the proliferation of LNCaP cells. However, the dose-response effect was only remarkable when the T concentration differences were between 1000 units. (see Figure 2).
On the other hand, 1 nM of DHT (Figure 2) significantly increased these cells’ viability compared with the vehicle-treated control (V) at 24 h. However, higher concentrations of DHT decreased cell proliferation. These data are according to the previously publicized by Horoszewicz et al., 1983. [31]
Results also indicated that after 48 and 72 hours of the T or DHT treatment, there were no significant differences in the number of viable LNCaP cells compared to vehicle-treated cells (data not shown). These facts could be explained because both steroids undergo glucuronidation reactions inside the LNCaP cells after 24 hours. [16, 32, 33] Due to these results, the following experiments were performed for 24 h. in the presence of 1 μM T and 1 nM DHT.
 |
Figure 2: Effect of different concentrations of testosterone (T) or dihydrotestosterone (DHT) on the proliferation of LNCaP cells ± standard error, measured at 24 h of treatment. |
*Significant difference (P<0.05) vs V
Antiproliferative activity of 2a-b derivatives in LNCaP cells
The effect of 2a-b derivatives on LNCaP cell proliferation is shown in Figure 3. Statistical analysis indicated that there was no significant difference (P> 0.05) vs. T, using 1 μM T plus 2a [1 nM and 1 μM] or 2b [0.1 nM, 1 nM, and 1 μM] in the incubation medium. Interestingly, steroid 2a at a 0.1 nM concentration significantly decreased the number of viable cells. (P<0.05).
However, FIN [15, 16, 34], in combination with T, showed no effect vs. T (P>0.05) on LNCaP cell proliferation, Figure 3. Lazier et al. previously reported that FIN did not induce the death of LNCaP cells. In contrast, dutasteride, a dual inhibitor of SRD5A1 and SRD5A2 activity, resulted in significant death of these cells at a concentration of 50 μM. [35] In addition, the LNCaP cell line does not express SRD5A2 [16], which explains the behavior of FIN in LNCaP cells in our experiments.
 |
Figure 3: The bars in the graph show the effect of testosterone (T) plus 2a or 2b on the proliferation of LNCaP cells ± standard error at 24 h of treatment. |
Antiproliferative activity of 3a-f derivatives in LNCaP cells
The antiproliferative activity of 3a-f steroids in combination with T is shown in Figure 4. Except for steroid 3b, all other series 3-compounds decreased the number of viable cells vs. T-treatment. The 3c derivative showed high dose-dependent activity, and 3d showed increased activity at all studied concentrations. However, 3a and 3f only showed activity at 0.1 nM and 1 nM vs. T concentrations.
 |
Figure 4: Effect of testosterone (T) plus 3a-f treatment on LNCaP cell proliferation ± standard error at 24 h. The compound 3c vs. |
DHT-dihydrotestosterone, V-vehicle, C-control without treatment
Antiproliferative activity of 4a-f derivatives in LNCaP cells
The proliferative activity of LNCaP cells treated with T or T plus 4 a-f ±standard error is shown in Figure 5. All products of series 4 decreased the number of viable cells, being 4f the one that showed the greatest potency and efficiency.
 |
Figure 5: shows the antiproliferative action of steroids 4a-f vs. T in the LNCaP cell line± standard deviation at 24 hours of treatment. |
The * on the bars indicates a significant difference vs. T (P <0.05).
Control (C); vehicle (V), testosterone (T), dihydrotestosterone (DHT).
Effect of different concentrations of 2a on the progress of the conversion of labeled T to DHT
Because steroid 2a at a 0.1 nM concentration (see 3.2 section) significantly decreased the number of viable cells. (P<0.05) compared with 1 nM and 1 μM concentrations, which did not show this effect, we determined the conversion of T to DHT in the presence of 0.1 nM, 1 nM, and 1 μM of 2a to explain these controversial results.
The optimal activity time of SRD5A1, obtained from the RS of LNCaP cells, was determined from the graph in Figure 6 and was 17 minutes.
 |
Figure 6: Optimal time of conversion of T to DHT in the presence of 30 μg of RS protein. This graph showed that solubilized microsomes of LNCaP cells converted T to DHT in an optimal time of 17 minutes. |
Figure 7 shows the T-to-DHT conversion rate in the presence of the LNCaP cells RS fraction. The maximum production of DHT turned out to be 368 ng/mg protein/h. These results are inconsistent with those previously published by Wu et al., who did not detect any rate conversion to DHT from 1 nM T in LNCaP cells using the mass spectrometry technique.
Furthermore, our results showed that the activity of SRD5A1 could be inhibited in the presence of 1 nM and 1 μM concentrations of 2a. Interestingly, in the presence of 0.1 nM of 2a, the conversion of T to DHT was not inhibited (Figure 7).
Contrary to expectations, the same amount of DHT was formed with or without the inhibitor (368 ng/mg protein/h). These data suggest that, at the 0.1 nM concentration, 2a acted as a positive cooperator for SRD5A1. The existence of heterotropic positive allosteric cooperators has been previously reported for cytochrome P-450. [36-38]
Harlow et al., 1997, demonstrated that progesterone underwent a hydroxylation reaction in a range of α-NF concentrations between 0–150 μM. While at higher concentrations of α-NF, the response did not occur. These data indicated that the group enzymes of the P-450 family have allosteric sites that can act as positive or negative cooperators depending on the concentration of the substrate.
In this sense, our group demonstrated [39] that the activity of SRD5A2 (obtained from prostate nuclear membrane fractions) increased in the presence of its reaction products, such as DHT or 5α-androstanedione. These results evidenced the presence of allosteric sites in this enzyme, which can act as positive cooperators.
 |
Figure 7: Effect of 2a on converting rate of T to DHT, using the solubilized microsomal fraction of LNCaP cells as a source of SRD5A1. |
Competition of series 1,2 and 3 derivatives for androgen receptor (AR) binding sites
IC50 values for series 1, 2, and 3 derivatives were determined according to the plots of concentration vs. percent binding of AR, as exemplified in Figure 8 for 4a-4f. The calculated IC50 and RBA values for MIB were 3.9 nM and 99%, respectively. In comparison, none of the derivatives of the other two series could displace the [3H]MIB of RA.
 |
Figure 8: Binding-competitive analysis by the androgen receptor (AR) present in the cytosolic fraction of the rat prostate. |
The results indicated that 4a-f did not bind to AR in rat prostate cytosol. However, it is necessary to consider that the AR of LNCaP cells displays a mutation in the ligand-binding domain, known as T877A. This mutation could affect the binding affinity of several of the hormonal or antihormonal compounds to AR. [40, 41]
Conclusions
This study showed that most of the derivatives of dehydroepiandrosterone (2a-b, 3a-f, and 4a-f) decreased the number of viable LNCaP cells in the presence of T. While T and DHT increased the proliferation of these cells. However, FIN failed to decrease the number of viable cells under experimental conditions. 2a-b, 3a-f and 4a-f were previously identified by our group as SRD5A1 inhibitors. [22, 23]
Of the three studied series, the 4a-f derivatives showed the highest efficacy and potency in inhibiting the proliferation of LNCaP cells. These steroids did not bind to AR present in rat prostate cytosol.
The microsomal fraction of LNCaP cells could convert T into DHT in the presence of NADPH at 17 minutes of incubation at 37 °C, indicating the activity of SRD5A1. [27] In addition, 2a at concentrations of 1 nM and 1 μMblocked the conversion of T to DHT. However, in the presence of 0.1 nM of 2a, DHT production was the same as in the absence of the inhibitor. This result suggests the allosteric nature of SRD5A1. [38]
Previous pharmacological studies carried out by our group in hamsters showed that the derivatives of the three studied series did not cause toxicity during the six days of treatment. [22, 23] These steroids could potentially ameliorate metastatic prostate tumors.
Acknowledgment
This research received no external funding.
Conflict of Interest
There are no conflict of interest.
References
- Koutsilieris, M. Osteoblastic metastasis in advanced prostate cancer. Anticancer. Res. 1993, 13 (2), 443-449. From NLM.
- Koutsilieris, M. Skeletal metastases in advanced prostate cancer: cell biology and therapy. Crit. Rev. Oncol. Hematol. 1995, 18 (1), 51-64. DOI: 10.1016/1040-8428(94)00122-a From NLM.
- Msaouel, P.; Nandikolla, G.; Pneumaticos, S. G.; Koutsilieris, M. Bone microenvironment-targeted manipulations for treating osteoblastic metastasis in castration-resistant prostate cancer. Expert Opin. Investig. Drugs 2013, 22 (11), 1385-1400. DOI: 10.1517/13543784.2013.824422 From NLM.
- Achbarou, A.; Kaiser, S.; Tremblay, G.; Ste-Marie, L. G.; Brodt, P.; Goltzman, D.; Rabbani, S. A. Urokinase overproduction results in increased skeletal metastasis by prostate cancer cells in vivo. Cancer Res. 1994, 54 (9), 2372-2377. From NLM.
- Montgomery, R. B.; Mostaghel, E. A.; Vessella, R.; Hess, D. L.; Kalhorn, R. F.; Higano, C. S. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008, 68, 4447-4454, 10.1158/0008-5472.CAN-08-0249.
- Dehm, S. M.; Tindall, D. J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183-R196, 10.1530/ERC-11-0141.
- Dehm, S. M.; Tindall, D. J. Androgen receptor structural and functional elements: role and regulation in prostate cancer. Mol. Endocrinol. 2007, 21 (12), 2855-2863. DOI: 10.1210/me.2007-0223 From NLM.
- Krueckl, S. L.; Sikes, R. A.; Edlund, N. M.; Bell, R. H.; Hurtado-Coll, A.; Fazli, L.; Gleave, M. E.; Cox, M. E. Increased insulin-like growth factor I receptor expression and signaling are components of androgen-independent progression in a lineage-derived prostate cancer progression model. Cancer Res. 2004, 64 (23), 8620-8629. DOI: 10.1158/0008-5472.can-04-2446 From NLM.
- Levina, E.; Chen, M.; Carkner, R.; Shtutman, M.; Buttyan, R. Paracrine Hedgehog increases the steroidogenic potential of prostate stromal cells in a Gli-dependent manner. Prostate 2012, 72 (8), 817-824. DOI: 10.1002/pros.21500 From NLM.
- Heemers, H. V.; Regan, K. M.; Dehm, S. M.; Tindall, D. J. Androgen induction of the androgen receptor coactivator four and a half LIM domain protein-2: evidence for a role for serum response factor in prostate cancer. Cancer Res. 2007, 67 (21), 10592-10599. DOI: 10.1158/0008-5472.can-07-1917 From NLM.
- Kashat, M.; Azzouz, L.; Sarkar, S. H.; Kong, D.; Li, Y.; Sarkar, F. H. Inactivation of AR and Notch-1 signaling by miR-34a attenuates prostate cancer aggressiveness. Am. J. Transl. Res. 2012, 4 (4), 432-442, from NLM.
- Bruchovsky, N.; Sadar, M. D.; Akakura, K.; Goldenberg, S. L.; Matsuoka, K.; Rennie, P. S. Characterization of 5α-reductase gene expression in stroma and epithelium of human prostate. The Journal of Steroid Biochemistry and Molecular Biology 1996, 59 (5), 397-404. DOI: https://doi.org/10.1016/S0960-0760(96)00125-2.
- Liao, G.; Chen, L.-Y.; Zhang, A.; Godavarthy, A.; Xia, F.; Ghosh, J. C.; Li, H.; Chen, J. D. Regulation of Androgen Receptor Activity by the Nuclear Receptor Corepressor SMRT. Journal of Biological Chemistry 2003, 278 (7), 5052-5061. DOI: 10.1074/jbc.M206374200.
- Anderson, K. M.; Liao, S. Selective retention of dihydrotestosterone by prostatic nuclei. Nature 1968, 219 (5151), 277-279. DOI: 10.1038/219277a0 From NLM.
- Jenkins, E. P.; Andersson, S.; Imperato-McGinley, J.; Wilson, J. D.; Russell, D. W. Genetic and pharmacological evidence for more than one human steroid 5α-reductase. The Journal of Clinical Investigation 1992, 89 (1), 293-300. DOI: 10.1172/JCI115574.
- Wu, Y.; Godoy, A.; Azzouni, F.; Wilton, J. H.; Ip, C.; Mohler, J. L. Prostate cancer cells differ in testosterone accumulation, dihydrotestosterone conversion, and androgen receptor signaling response to steroid 5α-reductase inhibitors. The Prostate 2013, 73 (13), 1470-1482, https://doi.org/10.1002/pros.22694. DOI: https://doi.org/10.1002/pros.22694 (accessed 2022/11/15).
- Russell, D. W.; Wilson, J. D. Steroid 5α-reductase: Two Genes/Two Enzymes. Annual Review of Biochemistry 1994, 63 (1), 25-61. DOI: 10.1146/annurev.bi.63.070194.000325.
- Span, P. N.; Benraad, T. J.; Sweep, C. G. J.; Smals, A. G. H. Kinetic analysis of steroid 5α-reductase activity at neutral pH in benign prostatic hyperplastic tissue: Evidence for type I isozyme activity in the human prostate. The Journal of Steroid Biochemistry and Molecular Biology 1996, 57 (1), 103-108. DOI: https://doi.org/10.1016/0960-0760(95)00167-0.
- Thomas, L. N.; Lazier, C. B.; Gupta, R.; Norman, R. W.; Troyer, D. A.; O’Brien, S. P.; Rittmaster, R. S. Differential alterations in 5α-reductase type 1 and type 2 levels during development and progression of prostate cancer. The Prostate 2005, 63 (3), 231-239. DOI: 10.1002/pros.20188.
- Thomas, L. N.; Douglas, R. C.; Rittmaster, R. S.; Too, C. K. L. Overexpression of 5α-reductase type 1 increases the sensitivity of prostate cancer cells to low concentrations of testosterone. The Prostate 2009, 69 (6), 595-602.
- Arellano, Y.; Bratoeff, E.; Garrido, M.; Soriano, J.; Heuze, Y.; Cabeza, M. New ester derivatives of dehydroepiandrosterone as 5α-reductase inhibitors. Steroids 2011, 76 (12), 1241-1246.
- Bratoeff, E.; Sánchez, A.; Arellano, Y.; Heuze, Y.; Soriano, J.; Cabeza, M. In vivo and in vitro effect of androstene derivatives as 5α-reductase type 1 enzyme inhibitors. Journal of Enzyme Inhibition and Medicinal Chemistry 2013, 28 (6), 1247-1254. DOI: 10.3109/14756366.2012.729827 (accessed 2013/12/19).
- Arellano, Y.; Bratoeff, E.; Heuze, Y.; Bravo, M.; Soriano, J.; Cabeza, M. Activity of steroid 4 and derivatives 4a–4f as inhibitors of the enzyme 5α-reductase 1. Bioorganic & Medicinal Chemistry 2018, 26 (14), 4058-4064. DOI: https://doi.org/10.1016/j.bmc.2018.06.030.
- Negri-Cesi, P.; Poletti, A.; Colciago, A.; Magni, P.; Martini, P.; Motta, M. Presence of 5α-Reductase isozymes and aromatase in human prostate cancer cells and in benign prostate hyperplastic tissue. The Prostate 1998, 34 (4), 283-291. DOI: 10.1002/(SICI)1097-0045(19980301)34:4<283::AID-PROS6>3.0.CO;2-I (accessed 2019/07/16).
- Liu, Y.; Peterson, D. A.; Kimura, H.; Schubert, D. Mechanism of Cellular 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) Reduction. Journal of Neurochemistry 1997, 69 (2), 581-593. DOI: 10.1046/j.1471-4159.1997.69020581.x (acccessed 2019/07/16).
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. Journal of Immunological Methods 1983, 65 (1), 55-63. DOI: https://doi.org/10.1016/0022-1759(83)90303-4.
- Levy, M. A.; Brandt, M.; Greway, A. T. Mechanistic studies with solubilized rat liver steroid 5α-reductase: elucidation of the kinetic mechanism. Biochemistry 1990, 29 (11), 2808-2815. DOI: 10.1021/bi00463a025 (accessed 2013/11/04).
- Hendry Iii, W. J.; Danzo, B. J. Structural conversion of cytosolic steroid receptors by an age-dependent epididymal protease. Journal of Steroid Biochemistry 1985, 23 (6, Part 1), 883-893. DOI: http://dx.doi.org/10.1016/0022-4731(85)90043-3.
- Cabeza, M. Molecular interactions of levonorgestrel and its 5α-reduced derivative with androgen receptors in hamster flanking organs. Steroids 1995, 60 (9), 630-635.
- Liao, S.; Witte, D.; Schilling, K.; Chawnshang, C. The use of a hydroxylapatite-filter steroid receptor assay method in the study of the modulation of androgen receptor interaction. Journal of Steroid Biochemistry 1984, 20 (1), 11-17. DOI: https://doi.org/10.1016/0022-4731(84)90182-1.
- Horoszewicz, J. S.; Leong, S. S.; Kawinski, E.; Karr, J. P.; Rosenthal, H.; Chu, T. M.; Mirand, E. A.; Murphy, G. P. LNCaP Model of Human Prostatic Carcinoma1. Cancer Research 1983, 43 (4), 1809-1818. (acccessed 5/17/2023).
- Zhou, J.; Tracy, T. S.; Remmel, R. P. Glucuronidation of dihydrotestosterone and trans-androsterone by recombinant UDP-glucuronosyltransferase (UGT) 1A4: evidence for multiple UGT1A4 aglycone binding sites. Drug Metab. Dispos. 2010, 38 (3), 431-440. DOI: 10.1124/dmd.109.028712 From NLM.
- Bélanger, G.; Beaulieu, M.; Marcotte, B.; Lévesque, E.; Guillemette, C.; Hum, D. W.; Bélanger, A. Expression of transcripts encoding steroid UDP-glucuronosyltransferases in human prostate hyperplastic tissue and the LNCaP cell line. Mol Cell Endocrinol 1995, 113 (2), 165-173. DOI: 10.1016/0303-7207(95)03627-j From NLM.
- Faller, B.; Farley, D.; Nick, H. Finasteride: A slow-binding 5α-reductase inhibitor. Biochemistry 1993, 32 (21), 5705-5710. DOI: 10.1021/bi00072a028 (accessed 2013/04/05/).
- Lazier, C. B.; Thomas, L. N.; Douglas, R. C.; Vessey, J. P.; Rittmaster, R. S. Dutasteride, the dual 5α–reductase inhibitor, inhibits androgen action and promotes cell death in the LNCaP prostate cancer cell line. The Prostate 2004, 58 (2), 130-144, https://doi.org/10.1002/pros.10340. DOI https://doi.org/10.1002/pros.10340 (accessed 2022/12/01).
- Polic, V.; Auclair, K. Allosteric Activation of Cytochrome P450 3A4 via Progesterone Bioconjugation. Bioconjug. Chem. 2017, 28 (4), 885-889. DOI: 10.1021/acs.bioconjchem.6b00604 From NLM.
- Harlow, G. R.; Halpert, J. R. Analysis of human cytochrome P450 3A4 cooperativity: construction and characterization of a site-directed mutant that displays hyperbolic steroid hydroxylation kinetics. Proc Natl Acad Sci U S A 1998, 95 (12), 6636-6641. DOI: 10.1073/pnas.95.12.6636 From NLM.
- Harlow, G. R.; Halpert, J. R. Alanine-scanning Mutagenesis of a Putative Substrate Recognition Site in Human Cytochrome P450 3A4: Role of residues 210 and 211 in flavonoid activation and substrate specificity*. Journal of Biological Chemistry 1997, 272 (9), 5396-5402. DOI: https://doi.org/10.1074/jbc.272.9.5396.
- Cabeza, M.; Trejo, K. V.; Gonzlez, C.; Garca, P.; Soriano, J.; Heuze, Y.; Bratoeff, E. Steroidal 5α-reductase inhibitors using 4-androstenedione as substrate. Journal of Enzyme Inhibition and Medicinal Chemistry 2011, 26 (5), 712-719.
- Veldscholte, J.; Berrevoets, C. A.; Ris-Stalpers, C.; Kuiper, G. G. J. M.; Jenster, G.; Trapman, J.; Brinkmann, A. O.; Mulder, E. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. The Journal of Steroid Biochemistry and Molecular Biology 1992, 41 (3), 665-669. DOI: https://doi.org/10.1016/0960-0760(92)90401-4.
- Veldscholte, J.; Ris-Stalpers, C.; Kuiper, G. G. J. M.; Jenster, G.; Berrevoets, C.; Claassen, E.; van Rooij, H. C. J.; Trapman, J.; Brinkmann, A. O.; Mulder, E. A mutation in the ligand binding domain of the androgen receptor of human INCaP cells affects steroid binding characteristics and response to anti-androgens. Biochemical and Biophysical Research Communications 1990, 173 (2), 534-540. DOI: https://doi.org/10.1016/S0006-291X(05)80067- *https://gco.iarc.fr/:Global Cancer Observatory, World Health Organization. International Agency for Research on Cancer.

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


