Synthesis and Design of Novel Morpholinyl Mannich Bases for Potential Inhibitory Activity of SARS-Cov-2 Main Protease
Mohamed R. Elamin1* , Sondos Abdullah J Almahmoud1, Tarek A. Yousef1,2 , Ibrahim K. Farh3, Hajo Idriss4,5 and Amin Osman Elzupir1,5
, Sondos Abdullah J Almahmoud1, Tarek A. Yousef1,2 , Ibrahim K. Farh3, Hajo Idriss4,5 and Amin Osman Elzupir1,5
1Department of Chemistry, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh 11623, Saudi Arabia.
2Toxic and Narcotic drug, Forensic Medicine Department, Mansoura Laboratory, Medicolegal Organization, Ministry of Justice, P.O. Box 12432, Cairo 11435, Egypt.
3Department of Pharmaceutical Sciences, College of Pharmacy, King Saud Bin Abdulaziz University for Health Sciences (KSAU-HS),Riyadh, Saudi Arabia.
4Department of Physics, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh 11623, Saudi Arabia.
5Deanship of Scientific Research, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh 11623, Saudi Arabia.
Corresponding Author E-mail: sumanswami1994@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/390207
Article Received on : 02 Mar 2023
Article Accepted on : 15 Apr 2023
Article Published : 27 Apr 2023
Reviewed by: Dr. Rama Krishna
Second Review by: Dr. Paras Nath YADAV
Final Approval by: Dr. Pounraj Thanasekaran
In this study, a new mannich base 1-(2H-1,3-benzodioxol-5-yl)-3-(morpholin-4-yl)propan-1-one (Mor) was successfully prepared in good yield. The structure of the title compound was elucidated by 1H-NMR, FT-IR, UV-Vis and electron-impact mass spectroscopy. The inhibitory activity of Mor against SARS-CoV-2 main protease (Mpro) was investigated by means of molecular docking approach. Mor showed excellent binding affinity to the active residues of Mpro with low binding score energy. Further improvements in the results were obtained by four designated analogues to Mor, characterized by the introduction of different methoxyl and hydroxyl substituents. The hydroxyl groups in Mor analogues significantly improve the binding affinity to the active site of Mpro to 56%, the binding energy to -6.3 kcal/mol, as well as the
ability to form hydrogen bonds compared with nirmatrelvir as the reference Mpro inhibitor.
Docking; Morpholine; Mannich bases; Morpholine; SARS-CoV- main protease
Download this article as:| Copy the following to cite this article: Elamin M. R, Almahmoud S. A. J, Yousef T. A, Farh I. K, Idriss H, Elzupir A. O. Synthesis and Design of Novel Morpholinyl Mannich Bases for Potential Inhibitory Activity of SARS-Cov-2 Main Protease. Orient J Chem 2023;39(2). |
| Copy the following to cite this URL: Elamin M. R, Almahmoud S. A. J, Yousef T. A, Farh I. K, Idriss H, Elzupir A. O. Synthesis and Design of Novel Morpholinyl Mannich Bases for Potential Inhibitory Activity of SARS-Cov-2 Main Protease. Orient J Chem 2023;39(2). Available from: https://bit.ly/41OC72k |
Introduction
Modern medicinal chemistry includes research on novel antiviral medicines as one of its top focus areas1. Because viral infections are prevalent, and new hazardous viral diseases are developing due to pathogenic strains such as Coronaviruses and influenza viruses2. There has been a severe outbreak of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in China since the end of 2019, prompting a global response and posing a severe threat to public health. Currently, there is a limited treatment for COVID-19, which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Therefore, identifying potential pharmacological treatments for this disorder is imperative.
Nitrogen-containing heterocycles ((N- heterocycles) received considerable attention due to their significant therapeutic efficacy4. Heterocycles containing nitrogen and their synthetic analogs may produce new medications. Due to their interesting biological properties, these elements are widely used in natural or artificial biological processes. 5. These compounds possess a wide variety of chemical structures with anti-inflammatory, anti-cancer and anti-viral properties, low toxicity, and the potential to influence multiple cellular targets. 6. Some N-heterocycles found in plants, have long been used as phytochemical medicines7. N-heterocycles are crucial for biochemical reactions. biochemical processes in living cells and their widespread distribution in natural products8. The primary ingredients of most enzymes consist of aromatic heterocycles, while the primary ingredients of most coenzymes are non-amino acids9. The most important nitrogen-containing heterocyclic compounds have numerous biological targets with their structural and pharmacological properties10.
Among many N-containing heterocycles, morpholine derivatives have attracted considerable attention due to their biological actions, which can be found in various therapeutic fields.
Morpholines can be considered a fundamental class and essential component in chemical synthesis, and many morpholine derivatives have attracted attention due to their unique and diverse uses. It is frequently chosen as a precursor material for synthesizing enantiomerically pure -amino acids, -amino alcohols, and peptides12. The popularity of the morpholine moiety can be attributed to many reasons. For instance, the oxygen atom at the center of morpholine might participate in donor-acceptor-type interactions with the appropriate receptor, increasing binding affinity. Moreover, the electronegative effect of oxygen atom’s decreases the nitrogen atom’s basicity13. In addition, morpholine derivatives are one of the structural components of many natural sources, 14, such as alkaloids, polygonapholine, chelonian, antioxidant and chelonian15,16.
Chelonian showed antibacterial action against Bacillus subtilis and an anti-inflammatory impact, whereas the two novel spiro alkaloids demonstrated antioxidant characteristics17. Due to the aforementioned biological aspects of morpholine, the current study aims to synthesize new morpholinyl mannich base and investigate their capability to inhibit the main protease of SARS- CoV-2 via molecular docking studies.
Materials and Methods
Synthesis of 1-(2H-1,3-benzodioxol-5-yl)-3-(morpholin-4-yl)propan-1-one (Mor) Morpholine (1.74 ml, 2.00 mmol), paraformaldehyde (612 mg, 2.00 mmol) and hydrochloric acid (few drops) were dissolved in dimethyl sulfoxide (40 ml) and the mixture was stirred at 100 °C for 12 hours. Then 3,4-(methylenedioxy) acetophenone (3.35 g, 2.00 mmol) was added to the mixture, and the pH was adjusted to around 2-3. The reaction was monitored by RP-HPLC-UV to completeness. The reaction mixture was allowed to cool to room temperature, neutralized to pH 7 using concentrated NaOH, then poured into distilled water (60 mL). The formed greyish yellow precipitate was collected by filtration, washed with acetonitrile, and left to dry in air to give the title compound (yield 78 %). 1H NMR (500 MHz, CDCl3) δ 7.56 (dd, J = 8.2, 1.7 Hz, 1H), 7.43 (d, J = 1.7 Hz, 1H), 6.85 (d, J = 8.2 Hz, 1H), 6.05 (s, 2H), 3.71 (t, J = 5.0 Hz, 2H), 3.10 (t, J =5.0 Hz, 2H), 2.81 (t, J = 5.0 Hz, 4H), 2.53 – 2.48 (m, 4H). FT-IR: 2895, 2801, 1661, 1588, 1422, 1250, 1217, 1013 cm−1; EI-MS m/z [M-H]+ 262 (requires for 263.1158 C14H17NO4).
Molecular docking
The ligands were prepared using ChemSketch in mol forms. Then converted to PDB forms with OpenBabel online tools converter, enabling add hydrogens and generate 3D coordinates functions. The reference, Nirmatrelvir, used in this study, and was obtained from PubChem library. Their structures energies were minimized utilizing the Molecular Modeling Toolkit plugin UCSF Chimera software, attempting 5000 steepest descent steps of 0.02 Å and 5000 conjugate gradient steps of 0.02 Å. The charges were assigned using antechamber. The crystal structure of the 3D Mpro was taken from the Protein Data Bank (PDB ID: 6Y2E). In order to prepare it for docking analysis, water residues have been removed, the missed hydrogens were added, and net charge was computed using antechamber [36]. Then its energy was minimized using the same tools and protocols used for ligands, utilizing 1000 steepest descent steps and 20 conjugate gradient steps. Molecular docking was achieved out with AutoDock Vina plugin UCSF Chimera software. The grid box size used was (32.0, 61.0, 60.0) Å, centered at (–17.0 × -25.0 × 16.0) Å. Conformers images interaction with the active sites of Mpro of SARS-CoV-2 were visualized and processed via UCSF Chimera software18-24.
Results and discussion
Synthesis and characterization
The target compound (MOR) was synthesized using a reported procedure and the synthetic route is illustrated in Scheme 1. Compound MOR was synthesized be dissolving morpholine and paraformaldehyde in DMSO and after adjusting the PH of the solution to around 2-3, acetophenone derivative was added and the target compound was obtained in a good yield.
 |
Scheme 1: Synthetics route of Mor. |
 |
Figure 1: 1H-NMR spectra of Mor. |
The structure of the final compound was characterized by 1H-NMR, FT-IR and mass spectroscopy. In 1H-NMR (Figure 1), the disappearance of the singlet peak around 2.53 ppm which corresponds to the CH3 group in the starting materials material ( 3,4-(Methylenedioxy)acetophenone)25 along with the appearance of the triplet peak at 3.71 ppm indicate a successful coupling. FT-IR spectrum (Figure 2) did not show any signs of the presence of NH stretch bands in the range 3310-3350 cm−1 which indicates the formation of C-N bond. The UV-vis absorption spectrum of MOR is presented in Figure 3, and was carried out in methanol solution (2 ppm). The UV-Vis absorption spectrum of MOR shows a wide absorption band in the UV region from 200 nm to 400 nm with four maxima of absorption (λmax) at 237, 262, 309 and 315 nm. Figure 4 showed the mass spectra of MOR using electron impact technique. The present of the parent molecular peak with the mass of 262 for m/z [M-H]+ was detected.
 |
Figure 2: FT–IR spectra of Mor. |
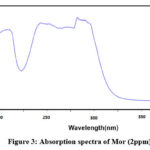 |
Figure 3: Absorption spectra of Mor (2ppm). |
 |
Figure 4: Mass spectra of Mor. |
Docking Analysis
MOR ligand was docked to the active site of Mpro with promising binding energy ranged from – to -5.9 kcal/mol. The lower bound RMSD is 0.00 and upper bound is 6.96, indicating stable and good binding affinity to Mpro receptor. The binding affinity or the preferences of the conformers to be docked to the active site was 44%. To test the efficiency of the docking investigation, Nirmatrelvir was docked to the Mpro as a reference. Nirmatrelvir was thoroughly studied as Mpro inhibitor25-27. Its shows binding affinity of 11% to the active pocket, with binding energy of 6.5 kcal/mol and RMSD ranged from 22.535 to 26.1. Nirmatrelvir do not show ahydrogen bond to GLU 166 residue as well as catalytic dyad of HIS 41 and CYS 145, its form hydrogen bond to CYS 44 (Figure5).
 |
Figure 5: (a) Mor in complex form with SARS-CoV-2 main protease with particular attention to the active residues of GLU 166, CYS 145, HIS 41. (b) The reference of Nirmatrelvir/Mpro complexes. |
To further improve these results four compounds were designed, introducing the different substituents to phenyl group (scheme 2). The full results were listed in Table 1. The replacement of methoxy groups with hydroxyl groups significantly improve the efficiency of the docking by increasing the binding affinity to the active site of Mpro, score energy, as well as the ability to form hydrogen bonds (Figure 6).
 |
Scheme 2: Chemical structures of synthesized MOR, its designed analogues, and the reference compound nirmatrelvir. |
 |
Figure 6: (a) 1MeO in complex form with SARS-CoV-2 main protease with particular attention to the active residues of GLU 166, CYS 145, HIS 41. (b) 1OH/Mpro complexes. (c) diMeO/Mpro complexes. (d) diOH/Mpro complexes. |
Table 1: The binding affinities the synthesized Mor, its designed analogues, and the reference compound nirmatrelvir with main protease of SARS-CoV-2 (Mpro).
|
ID |
Binding affinity |
Score Kcal/mol |
RMSD |
Hydrogen bonding |
van der Waals |
|
Mor |
44% |
-5.5 to -5.9 |
0.0 to 6.96 |
– |
HIS 163, MET 49, HIS 41, SER 46, GLU 166, THR 45,
ASN 142, THR 25, CYS 145, THR 24, CYS 44, PHE
140, MET 165, LEU 141, HIS 164 |
|
diOH |
56% |
-5.5 to -6.3 |
0.0 to 5.99 |
CYS 145, HIS 163,
GLU 166, LEU
141, SER 144, PHE
140 |
LEU 141, HIS 163, PHE 140, GLU 166, SER 144, MET
165, MET 49, CYS 145, ASN 142, HIS 41, HIS 164,
GLY 143, THR 25, LEU 27 |
|
diMeO |
11% |
-5.4 |
27.28 |
– |
MET 49, THR 25, LEU 27, GLU 166, HIS 163, HIS 41,
ASN 142, LEU 141, CYS 145, SER 144, PHE 140 |
|
1OH |
44% |
-5.4 to -5.5 |
|
HIS 163, GLU 166,
LEU 141 |
SER 144, ASN 142, LEU 141, PHE 140, HIS 163, GLU
166, MET 165, THR 25, HIS 41, CYS 44, CYS 145,
HIS 41, THR 25 |
|
1MeO |
11% |
-5.5 |
28.87 |
– |
MET 165, GLU 166, HIS 163, CYS 44, MET 165, CYS
145, MET 49, THR 25, SER 46, GLN 189, THR 45, HIS164, HIS 41, GLN 189 |
|
Nirmatrelvir |
11% |
-6.5 |
22.54 |
CYS 44 |
GLN 189, GLU 166, ASN 142, CYS 145, CYS 44, MET
49, THR 25, HIS 41, LEU 141, SER 144, HIS 163, LEU
167, PHE 140, PRO 168 |
All ligands and nirmatrelvir reference were interacted to the catalytic dyed of HIS 41 and CYS 145 via van der Waals forces. The compound diOH show high ability to form hydrogen bonds with several residues including CYS 145 and GLU 166, with RMSD significantly higher than that reported for nirmatrelvir, and comparable binding score energy up to 6.3 kcal/mol. The carbonyl group in diOH form hydrogen bond to the important residue GLU 166 with a distance of 2.195 Å. Further, the binding affinity to the Mpro active site is 56%.
Conclusion
In conclusion, this work presents the synthesis of the ligand Mor, which was successfully synthesized and characterized by FT-IR, NMR, and EI-MS, and UV-Vis. Analogous ligands with methoxy and hydroxy groups were designed and their activity against Mpro was investigated by means of molecular docking approach. The designed and synthesized ligands showed excellent binding affinity to the active residues of Mpro with low binding energy. Our results suggest that these ligands may be potential candidates for SARS-CoV-2 Mpro, especially the compound diOH, and thus further studies in vitro and in vivo are recommended.
Acknowledgment
This research was supported by the Deanship of Scientific Research, Imam Mohammad Ibn Saud Islamic University (IMSIU), Saudi Arabia, Grant No. (21-13-18-046).
Conflicts of interest
The authors declare no conflict of interest.
References
- Wang Z, Cherukupalli S, Xie M, Wang W, Jiang X, Jia R, Pannecouque C, De Clercq E, Kang D, Zhan P, Liu X. J. Med. Chem. 2022. 17;65(5):3729-57.
CrossRef - Pantaleo G, Correia B, Fenwick C, Joo VS, Perez L. Nat. Rev. Drug Discovery. 2022 Sep;21(9):676-96.
- Li CX, Noreen S, Zhang LX, Saeed M, Wu PF, Ijaz M, Dai DF, Maqbool I, Madni A, Akram F, Naveed M. Biomed. Pharmacother. 2022 .1;146:112550.
CrossRef - Jiang J, An Z, Li M, Huo Y, Zhou Y, Xie J, He M. J. Environ. Chem. Eng. 2023 . 1;11(1):109193.
- Aatif M, Raza MA, Javed K, Nashre-ul-Islam SM, Farhan M, Alam MW. Antibiotics. 2022 ;11(12):1750.
- Łowicki D, Przybylski P. Eur. J. Med. Chem. 2022. 18:114303.
CrossRef - Gao B, Yang B, Feng X, Li C. Nat. Prod. Rep. 2022;39(1):139-62.
CrossRef - Insuasty D, Castillo J, Becerra D, Rojas H, Abonia R. Molecules. 2020 . 24;25(3):505.
CrossRef - Minhas R, Bansal Y, Bansal G. Med. Res. Rev. 2020;40(3):823-55.
CrossRef - Kerru N, Gummidi L, Maddila S, Gangu KK, Jonnalagadda SB. Molecules. 2020.20;25(8):1909.
CrossRef - Gattu R, Ramesh SS, Nadigar S, Ramesh S. Antibiotics. 2023 Mar 7;12(3):532.
CrossRef - Chen C, Guo SM, Sun Y, Li H, Hu N, Yao K, Ni H, Xia Z, Xu B, Xie X, Long YQ. Eur. J. Med. Chem. 2023 Mar 11:115267.
- S.M. Yang, R. Malaviya, L.J. Wilson, R. Argentieri, X. Chen, C. Yang, B. Wang, D. Cavender, W.V. Murray, Bioorg. Med. Chem. Lett. 2007, 17, 326
CrossRef - Wei JS, Yang S, Wei Y, Shamsaddinimotlagh S, Tavakol H, Shi M. Org. Chem. Front. 2023.
- L. Yurttaş, Z.A. Kaplancıkl, Y. Özkay, J. Enz. Inhib. Med. Chem, 2013, 5, 1040
CrossRef - H. Zhao, H. Thurkauf, K. Hodgetts, X. Zhang, Bioorg. Med. Chem. Lett, 2002, 10, 310
- M. Kimura, T. Masuda, K. Yamada, N. Kubota, N. Kawakatsu, M. Mitani, K. Kishii, M. Inazu, T. Namiki, Bioorg. Med. Chem. Lett, 2002, 12, 1947.
CrossRef - Wang, J.; Wang, W.; Kollman, P. A.; Case, D. A., Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, (2), 247-260.
CrossRef - Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Greenblatt, D. M.; Meng, E. C.; Ferrin, T. E., UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, (13), 1605-1612.
CrossRef - Elzupir, A. O. J. Mol. Struct. 2020, 1222, 128878.
CrossRef - Al-Janabi, A. S.; Elzupir, A. O.; Yousef, T. A. J. Mol. Struct. 2021, 1228, 129454.
CrossRef - O’Boyle, N. M.; Banck, M.; James, C. A.; Morley, C.; Vandermeersch, T.; Hutchison, G. R.. J. cheminformatics. 2011, 3, (1), 1-14.
CrossRef - Shapovalov, M. V.; Dunbrack Jr, R. L. Structure 2011, 19, (6), 844-858.
CrossRef - Trott, O.; Olson, A. J. J. Comput. Chem. 2010, 31, (2), 455-461.
- Umesha B.; and Basavaraju Y. B., Medicinal Chemistry Research, 2014, 23, 3744-3751.
CrossRef - Joyce, R. P., Hu, V. W., & Wang, J. 2022. Med. Chem. Res., 31(10), 1637-1646.
CrossRef - Chia, C. B., & See, Y. Y. Med. Chem. Res.2022. 13(9), 1388-1389.
CrossRef - Mótyán, J. A., Mahdi, M., Hoffka, G., & Tőzsér, J. Int. J. Mol. Sci. .2022. 23(7), 3507.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()



{kind=link}