Novel Process for Preparation of Oxazolonone, Benzoxazolonone.
Umesh K. Patil1 , Sayujjata R. Vaidya1 and Balaji R. Madje2*
, Sayujjata R. Vaidya1 and Balaji R. Madje2*
1Department of chemistry, Vivekanand Arts and S.D. Commerce and Science College, Samarth Nagar, Aurangabad, MS, India.
2Department of chemistry, Vasantrao Naik Mahavidyalaya, Aurangabad (MS) India.
Corresponding Author E-mail: drmadjebr@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/390126
Article Received on : 20 Oct 2022
Article Accepted on : 05 Feb 2023
Article Published : 21 Feb 2023
Reviewed by: Dr. Hemaprobha Saikia
Second Review by: Dr. Vipin A Nair
Final Approval by: Dr. Arpita Biswas
Synthesis of oxazolonone, benzoxazolononeby using amino hydroxy compound, alkyl haloformate, trialkylamine and DMF as solvents. Herein used simple carbonylation agent such as alkylhaloformate instead of phosgene and Triphosgene. Developed novel and simple synthesis method.
KEYWORDS:Alkylhaloformate; Benzoxazolonone; Oxazolo pyridinone; Oxazolonone
Download this article as:| Copy the following to cite this article: Patil U. K, Vaidya S. R, Madje B. R. Novel Process for Preparation of Oxazolonone, Benzoxazolonone. Orient J Chem 2023;39(1). |
| Copy the following to cite this URL: Patil U. K, Vaidya S. R, Madje B. R. Novel Process for Preparation of Oxazolonone, Benzoxazolonone. Orient J Chem 2023;39(1). Available from: https://bit.ly/3EnUC3A |
Introduction
The development of heterocycles compound like Oxazolonone, benzoxazolononeasframe, containing a high degree of variety has become a number one focus in medicine discovery. Variousmodifications on the ring by addition of various substituents suggestionnew products with advanced natural biographies. Considering strategic medical importance of Oxazolonone, benzoxazolonone, developed easy and possible novel synthesis technique.
As per literature oxazolo pyridinone synthesized by carbonylation of 2-amino-3-hydroxy pyridine using a) 1, 1-carbonyldiimidazole in THF1, 2, b) Triphosgene in toluene-chloroform3, c) carbon monoxide in dimethyl formamide4, d) carbon monoxide in presence of potassium iodide, palladium (II) iodide in autoclave5-6, e) N, N’-disuccinimidyl carbonate7- 9, f) urea 8, g) carbon disulphide in ethanol 10, h) hexamethylphosphoramide 11, i) phosgene 12.
Benzimidazolone synthesized by carbonylation of 1,2-diamino benzene using a) urea and DMF 13-14,b) carbon dioxide in water 15, c) Ι, Γ-carbonyldiimidazole (CDI) in DMF 16-18, ethyl acetate 19, in benzene 20, and water 21, d) using corbonmonoxide , 1-Methylpyrrolidine and selenium in tetrahydrofuran22, e) CO2; oxygen; KI; palladium (II) iodideand 1, 2-dimethoxyethane23, f) in 1-Methylpyrrolidine at 100 °C 24. Also phthalic anhydride treated withtrimethylsilylazide in tetrahydrofuran for 30h; Curtius rearrangement with sodium azide; acetic acid 25-26. Formylation of O–nitrophenols or O–nitroaniline and selenium as catalyst 27. From amides by iodosylbenzene-induced Hofmann rearrangement28. Lanthanide-catalysed cyclocarbonylation and cyclothiocarbonylation 29.
Main drawback of above all route of synthesis is generation of toxic reaction waste and critical handling of phosgene and triphosgene. In present invention, we developed simple commercially viable, non-phosgene, synthetic method.
Experimental
Solvents and key raw materials used for this experimentation,bought from commercial sources, and used as such. Melting points analysed on Electro-thermal IA 9100 apparatus (Shimadzu), 1H NMR spectra analysed on a Bruker (400 MHz) spectrometer in DMSO-d6. The chemical shifts as ppm against tetramethylsilane (TMS)as internal reference. Duringreaction, the formation of compounds, checked by TLC on silica gel plates of 0.5mm thickness and checked location of spots by iodineand UV light.
Common procedure for preparation of compound(3a-e).
Compound 1a-e (1eq), compound-2 (1.05eq) and DMF (6.38V) taken in round bottom flask. Added Trialkylamine (1.50eq) in 1h. Heated reaction mass at 75-80°C, 4h. Heated the reaction mass at 105 to 110°C over a period of 10-15h. Distilled out DMF at 70 to 75°C using vacuum. Added water (8V) and distilled out traces of DMF. Cooled the reaction mass at 0 to 5°C and filtered. Wet cake dried at 55 to 60°C under vacuum. Obtained compound 3a-e (Scheme 1).
[ 1,3] Oxazolo [4, 5-b] pyridin-2-ol (3a).
Yield = 91%, Grey solid powder, Melting point is 212-214°C, 1HNMR400MHz, DMSO (D6): ppm 7.081-7.114(d, 1H, J=13.2Hz), 7.615-7.638 (d, 1H, J= 9.2Hz), 8.018-8.034 (d, 1H, J=6.4Hz ),12.438(s, 1H-N) .13CNMR 400MHz, DMSO (D6): ppm 116,117.89,137.48,142.5, 146.34,153.48.
6-chloro-3H-oxazolo[4,5-b] pyridin-2-one(3b),
Yield = 91%, Whitepowder, Melting point is 183-185°C, 1H NMR 400 MHz, DMSO (D6): ppm 7.922-7.917(d, 1H, J= 2Hz),8.093-8.088(d,1H, J=2Hz),12.646(s,1H-N). 13C NMR 400MHz, DMSO (D6): ppm 116.64, 124.22, 137.62, 140.69, 145.22, 153.21.
1, 3-dihydro-2H-benzimidazol-2-one (3C)
Yield = 90%, White powder, Melting point is 318-320°C, 1H NMR 400 MHz, DMSO (D6): ppm,6.910(s, 4H,), 10.576 (s, 2H-N). 13C NMR 400MHz, DMSO (D6): ppm 108.45,120.37,129.65,155.24.
Results and Discussion
Carbonylation method is most efficient for synthesis of oxazolopyridine and benzoxazolones. The major advantage of this method is non-phosgene route of synthesis. Carbonylating agent is easily available and can be manage easily on commercial scale. Trialkylamine can be easily recover and recycle in the process which reduces production cost. Phosgene is very toxic and required special storage facility where as alkylhaloformate storage is not complicated as compared to phosgene and triphosgene.
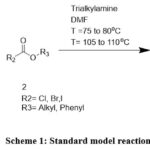 |
Scheme 1: Standard model reaction. |
Compound 1a-e treated with compound 2 and trialkylamine in DMF as a solvent. (Scheme-1), as per General procedure for preparation of compound (3a-e).and product 3a-e yield summarized in (table-1).
Table 1: Synthesis of compound 3a-e.
|
Entry |
Product |
Compund-1 |
Melting point °C |
%Yield. |
||||
|
R |
X |
Y |
Z |
Found |
Reported |
|
||
|
1 |
3a |
H |
N |
N |
O |
212-214 |
212-216 |
91 |
|
2 |
3b |
Cl |
N |
N |
O |
183-185 |
183-186 |
91 |
|
3 |
3c |
H |
C |
N |
N |
318-320 |
320-322 |
90 |
|
4 |
3d |
H |
C |
N |
O |
139-141 |
139-142 |
88 |
|
5 |
3e |
H |
C |
O |
O |
119-121 |
120-122 |
65 |
Screening of different solvents such as DMF, Methanol, Acetone, DCM, and Toluene, for synthesis of 3a. Reaction done as per general procedure for preparation of compound (3a-e). Due to reaction temperature condition lower boiling solvents are not suitable. Toluene and DMF used as a mixture by selecting different ratio, noticed in 100% toluene conversion is zero. We studied increasing DMF composition observed increasing yield of reaction and in 100% DMF obtained best yield. Commercial point of view single solvent is good for cost effective process. We found that DMF is best solvent for reaction as reactants and intermediate having good solubility for effective interaction. We observed that desired product 3a formed with in 20h, with 91% yield. Yield of 3a formation mentioned in (table 2).
Table 2: Effect of solvent.
|
Entry |
Toluene % |
DMF % |
% Yield |
|
1 |
100 |
0 |
12 |
|
2 |
90 |
10 |
18 |
|
3 |
80 |
20 |
32 |
|
4 |
70 |
30 |
40 |
|
5 |
60 |
40 |
62 |
|
6 |
50 |
50 |
70 |
|
7 |
40 |
60 |
74 |
|
8 |
30 |
70 |
76 |
|
9 |
20 |
80 |
89 |
|
10 |
10 |
90 |
89 |
|
11 |
0 |
100 |
91 |
After screening of various carbonylation agent such as alkylhaloformate as per General procedure for preparation of compound (3a-e).we noticed that ethyl and methyl chloroformate gives 91% yield of compound 3a. Yield of compound 3a summarized in (Table 3).
Table 3: Optimization of alkylhaloformate for synthesis of compound-3a.
|
Entry |
Alkylhaloformate |
% Yield |
|
1 |
Ethyl chloroformate. |
91 |
|
2 |
Methyl chloroformate. |
91 |
|
3 |
N, N-Dimethyl carbamyl chloride. |
82 |
|
4 |
Propyl chloroformate |
79 |
|
5 |
Butyl chloroformate. |
75 |
|
6 |
Phenyl chloroformate. |
68 |
After screening of various trialkylamine as per general procedure for preparation of compound (3a-e). we noticed that triethylamine and N, N-diisopropyl ethylamine gives 91% yield of compound 3a. Yield of compound 3a summarized in (Table 4)
Table 4: Optimization of Trialkylamine for synthesis of compound 3a.
|
Entry |
Trialkylamine |
Temperature |
% Yield |
|
1 |
TEA |
75 to 110°C |
91 |
|
2 |
N, N diisopropyl ethylamine |
75 to 110°C |
91 |
|
3 |
N, N dimethyl benzyl amine. |
75 to 110°C |
88 |
|
4 |
DMAP |
75 to 110°C |
88 |
|
5 |
Pyridine |
75 to 110°C |
85 |
Conclusion
We have developed non phosgene, and commercially viable synthetic method for synthesis of oxazolonone, benzoxazolonone. Important advantage of this method is clean reaction profile, high yield, simple carbonylating agent. Acid scavenger can be recover and recycle in the process.
Acknowledgement
Authors thankful to Principal, Vivekanand Arts & S.D. Commerce and Science College, Aurangabad (MS)-431001 for his encouragement and support.
Conflict of Interest
Authors does not have any conflict of interest.
References
- Affinium, P. I.WO2007/67416.2007, A2.
- Deau, E.; Robin, E.; Voinea, R.; Percina, N.; Satała, G.; Finaru, A.L.; Chartier, A.; Tamagnan, G.; Alagille, D.; Bojarski, A.J.; Morisset-Lopez, S.; Suzenet, F.; Guillaumet, G.J. Medi. Chem.2015, 58,8066 – 8096
CrossRef - Qian, J. Q.; Yan, P.C.; Che, D. Q.; Zhou, Q. L.;Li,Y, Q.Tetra. Lett.2014, 55, 1528 – 1531
CrossRef - Wang, X.; Ling, G.; Xue, Y.; Lu, S.European J Org Chem.2005, 8, 1675 – 1679
CrossRef - Mancuso R.; Raut, D.S.; Della, C.N.; Fini, F.; Carfagna, C.; Gabriele, B. Chem. Sus. Chem.2015,13, 2204 – 2211
CrossRef - Fraser.; Tittensor.J.Chem. So. 1957, 4625.
- Takeda, K.; Ogura, H.Synth.Communi.1982, 12, 213 – 218
CrossRef - Aliev, T.; Levkovich, A.; Kartstev.Chem.Hetero.Comp.1997,33,1337 – 1340
CrossRef - Takeda, K.; Tsuboyama, K.; Takayanagi, H.; Shirokami, R.; Takeura, M.; Ogura, H.; Chem. Pharma. bulletin.1989, 37, 2334 – 2337
CrossRef - Mincheva, Z.; Courtois, M.; Andreu, F.; Rideau, M.; Viaud-Massuard, M. Phytochemistry. 2005, 66, 1797 – 1803
CrossRef - Aliev, L.; Kristallovich.; Abdullaev.; Kartsev. Chem. Hetero. Comp.1999, 35, 84 – 86
CrossRef - Ruefenacht, K. K. Hel. Chimi. Acta.1976,59, 1593 – 1612
CrossRef - Dubey, P. K; Naidu, A; Anandam, V; Hemasunder, G; Ind.J.Chem.Org.Med.Chem.2005, 44,1239 – 1242
- Rekunge, D. S; Khatri, C. K; Chaturbhuj; G. U. Tetra. Letters. 2017, 58: 4304 – 4307.
CrossRef - He, Z.; Kang, X.; Sadeghzadeh, S.M.; Yu, P.; Zhang,H.; Zhang, Y.; Zhao, Y. Cat. Lett. 2020
- Agrahari, A. K.; Singh, A. S; Singh, S. K.; Tiwari, V. K.; Yadav, M. S. Syn. 2019, 51, 3443 – 3450
CrossRef - Kilchmann, F.; Marcaida, M. J.; Kotak, S.; Schick, T.; Boss, S.D.; Awale, M.; Gönczy, P.; Reymond, J.L. J. Med. Chem.2016, 59,7188 – 7211
CrossRef - Velagapudi, U. K.; Langelier, M. F.; Delgado-Martin, C.; Diolaiti, M. E. Bakker, S.; Ashworth, A.; Patel, B. A.; Shao,X.;Pascal, J.M.; Talele, T.T.J. Med.Chem,2019, 62, 5330 – 5357.
CrossRef - Kornberg, B.E.;Lewthwaite, R.A.; Manning, D.; Nikam, S.S.; Scott, I.L. US2003/18021, 2003, A1
- Lellmann.; Wuerthner.; Jus. Lieb.Anna. der Chem,1885, 228, 229
CrossRef - Pellizzari. Gaz.Chi. Ital.1919,49, 22
CrossRef - Yoshida, T.; Kambe, N.; Murai,S.; Sonoda, N. Bull.Chem. So, Japan,1987, 60, 1793 – 1800
CrossRef - Gabriele, B.; Salerno, G.; Mancuso, R.; Costa, M. J. Orga. Chem.2004,69: 14,4741 – 4750
CrossRef - Yoshida, T.; Kambe, N.; Ogawa, A.; Sonoda, N. Phos.Sul.Rel. Elem. 1988, 38,137 – 148
CrossRef - López,H.S.; Enciso, J.E.; Ochoa-Teran, A.; Velazquez, J. I.; Sarmiento. J.Mend. Comm. 2016, 26: 69 – 71
CrossRef - Maffei.; Bettinetti.; Annali, d.C. 1959,49, 1809, 1812
- Wang, X.;Ling, G.;Xue, Y.;Lu, S. Euro. J.Org. Chem. 2005,8, 1675 – 1679
CrossRef - Liu,P.;Wang, Z.;Hu,X.Euro J. Org. Chem.2012, 10, 1994 – 2000
CrossRef - Jing, Y.;Liu,R.;Lin, Y.;Zhou, X.Sci. China. Chem.2014,57,1117 – 1125
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


