Calculation of Vibrational Energies of AlH2 Using U(2) Lie Algebraic Approach
P. Suneetha and J. Vijayasekhar*
and J. Vijayasekhar*
Department of Mathematics, GITAM (Deemed to be University), Hyderabad, India.
Corresponding Author E-mail: vijayjaliparthi@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/380531
Article Received on : 30 Jul 2022
Article Accepted on : 21 Oct 2022
Article Published : 26 Oct 2022
Reviewed by: Dr. B.V.S.N. Hari Prasad
Second Review by: Dr. Sreeram Venigalla
Final Approval by: Dr. B.K. Sharma
Using the U(2) Lie algebraic approach, it is determined that the vibrational energies of the aluminumdihydride (AlH2) molecule up to the fifth overtone and reported the combinational bands up to the third harmonic.In this approach, the algebraic parameters and operators are used to represent the Hamiltonian operator to calculate the vibrational energies.
KEYWORDS:aluminumdihydride; Hamiltonian operator; U(2) Lie algebras; Vibrational energies
Download this article as:| Copy the following to cite this article: Suneetha P, Vijayasekhar J. Calculation of Vibrational Energies of AlH2 using U(2) Lie Algebraic Approach. Orient J Chem 2022;38(5). |
| Copy the following to cite this URL: Suneetha P, Vijayasekhar J. Calculation of Vibrational Energies of AlH2 using U(2) Lie Algebraic Approach. Orient J Chem 2022;38(5). Available from: https://bit.ly/3f4bF1w |
Introduction
The interpretation and analysis of the molecular spectra of molecules is a fascinating research area of molecular physics. Vibrational energies have played anvital role in both theoretical and experimental techniques due to their diverse applications. Vibrational spectra is currently undergoing an exciting period of renewed attention, fuelled by the rapid development of sophisticated experimental techniques. Due to their numerous applications in the research study of vibrational energies of molecules, one and three dimensional [U(2), U(4)] Lie algebraic approaches have grabbed the attention of a larger scientific community. With the use of these approaches, one can easily obtain the vibrational and rotational degrees of freedom in a physical problems1. The vibrational energies of a molecule are represented by the eigenvalues of the Hamiltonian matrix. These approaches are successful in the research study of polyatomic molecules vibrational spectra 2-7. The vibrational and rotational energies are treated simultaneously in the U(4) Lie algebraic approach. When a molecule has more than four atoms, this approach becomes more complicated. To analyze the vibrational energies of aluminum dihydride, this limitation led us to utilize the U(2) Lie algebraic approach.
One dimensional Lie algebraic approach
A bent molecule with C2v point group symmetry is aluminium dihydride.This aluminium dihydride molecule contains the symmetry species, A1(Symmetric Stretch), B2 (Antisymmetric Stretch), and A1 (Bend).
For n vibrational modes, the general Hamiltonian 8-11 is
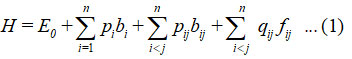
In equation (1), bi and bij are uncoupled and coupled bonds invariant operators, respectively, and known as
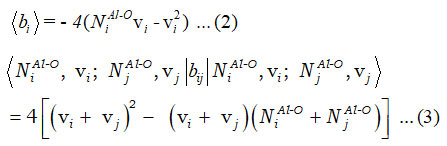
Majorana operator fij consist diagonal and non-diagonal matrix elements and it is useful to articulate the pair of local mode interactions 12.
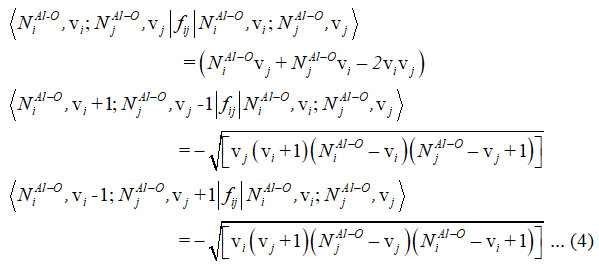
For two (Al-O) stretching vibrations of aluminumdihydride, the Hamiltonian operator written as
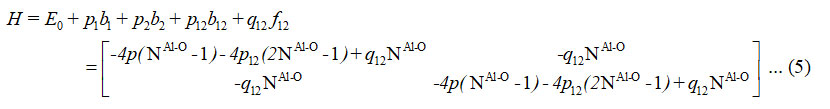
From spectroscopic data algebraic parameters P1 ,P2, P12 and q12 (cm-1) are determined . Two bonds (Al-O) are equivalent in the aluminum dihydride. As a result, we consider P1 = P2 = P and Vibron number, N1Al-o = N2Al-o= NAl-o in equations (2), (3) and (4).
Results and Discussion
The parameter p is determined using the energy equation (6)
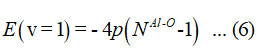
and the value of q12 calculated from the relation,
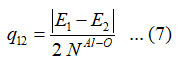
Where E1, E2 are the aluminum dihydride symmetric and antisymmetric vibrational energies, respectively. The number Ni for stretching bonds of aluminum dihydride is calculated from the relation,
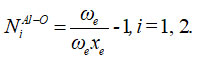
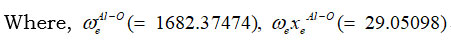
are correspondingly, vibrational harmonic and an harmonic spectroscopic constants 13.
The Lie algebraic approach is utilized to compute the vibrational energies of aluminium dihydride (in higher overtones and combinational bands), as indicated in the tables (1), (2) and (3).
Table 1: Vib. frequencies in fundamental mode (in cm-1).
|
Vib. Mode |
Symmetry |
Experimental# |
Computed |
|
v1 (Symmetric stretch) |
A1 |
1788 |
1788.38 |
|
v2 (bend) |
A1 |
760 |
758.7162 |
|
v3(antisymmetric stretch) |
B1 |
1828.6 |
1827.9732 |
#webbook.nist.gov/cgi/cbook.cgi?ID=C14457659&Units=SI&Mask=800#Electronic-Spec
Table 2: Vib. frequencies (overtone) (in cm-1)
|
Overtone |
Vib. mode |
||
|
Symmetric stretch (A1) |
Bend (A1) |
Antisymmetric stretch(B1) |
|
|
1 |
2v1(3563.761) |
2v2(1509.995) |
2v3(3644.028) |
|
2 |
3v1(5335.594) |
3v2(2237.022) |
3v3(5429.990) |
|
3 |
4v1(7091.096) |
4v2(2921.258) |
4v3(7258.733) |
|
4 |
5v1(8826.294) |
5v2(3620.721) |
5v3(8089.413) |
|
5 |
6v1(10622.643) |
6v2(4430.447) |
6v3(9758.083) |
Table 3: Combinational frequencies (in cm-1)
|
Combinational Band |
Computed |
Combinational Band |
Computed |
|
v1+ v2 |
2542.075 |
2v1+ v2 |
4317.456 |
|
v1+ v3 |
3615.685 |
v2+2v2 |
2258.336 |
|
v2 + v3 |
2580.668 |
v2 +2v3 |
4397.723 |
|
v1+2v1 |
5352.473 |
2v1+ v3 |
5391.066 |
|
v1+ 2v2 |
3293.354 |
v3+ 2v2 |
3331.947 |
|
v1+ 2v3 |
5432.740 |
v3+ 2v3 |
5471.333 |
Table 4: Parameters
| NAl-O(stretch) |
58 |
| NAl-O(stretch) |
38 |
| P(stretch) |
-7.9275 |
| P(bend) |
-5.1351 |
| P12(stretch) |
0.0415 |
| P13(bend) |
-1.2968 |
| q12(stretch) |
0.3327 |
| q13(bend) |
1.9621 |
Conclusion
We compared the examined data which was in the table (1) with the calculated fundamental vibrational energies of the aluminumdihydride.
Vibrational energies up to theharmonic level six and combinational bands upto the harmonic level three were described using the one dimensional [U(2)] Lie algebraic approach in tables (2) and (3).
Acknowledgment
The author, J. Vijayasekhar, would like to thanks GITAM(Deemed to be University), Vishakhapatnam, India, for providing financial assistance of this study under GITAM: Research seed Grants.
Conflicts of Interest
The authors declare no conflict of interest.
References:
- Iachello, F.; Oss, S.; Lemus, R.J. Mol. Spectrosc.1991, 149 (1), 132-151.
CrossRef - Karumuri, S.R.; Sravani, K.G.; Vijayasekhar, J.; Reddy, L.S.S. Acta Phys. Pol. A. 2012, 122 (1), 49 52.
CrossRef - Karumuri, S. R.;Srinivas, G.;Vijayasekhar, J.; Rao, V. U. M.;Srinivas, Y.; Sunil Babu, K.; Kumar, V. S. S.;Hanumaiah, A. Chin. Phy. B.2013, 22(9), 090304(1-8).
CrossRef - Iachello, F.; Oss, S.J. Mol. Spectrosc. 1992, 153 (1–2), 225-239.
CrossRef - Balla, M.R.; Vijayasekhar, J. Mol. Phys. 2021, 115 (5), e1828634.
CrossRef - Balla, M.R.; Vijayasekhar, J. PolycyclAromat Compd. 2021. 42(7), 4684-4699
CrossRef - Balla, M.R.; Venigalla, S.; Jaliparthi, V. Acta Phys. Pol. A. 2021, 140 (2), 138-140.
CrossRef - Oss, S. Adv. Chem. Phys.1996, 93, 455-649.
- Iachello, F.; Levine, R. D. Oxford University Press, Oxford.1995.
- Vijayasekhar, J.;Balla, M.R. Spectrochim. Acta A. 2022, 264, 120289(1-8).
CrossRef - Vijayasekhar,J.Ukr. J. Phys. Opt. 2022, 23(3), 126-132.
CrossRef - Vijayasekhar,J; Srinivas, N; Kumar, A. K. Orient. J. Chem. 2022, 38(1): 204-206.
CrossRef - Irikura, K.K. J. Phys. Chem. Ref. Data. 2007, 36(2), 389-398.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


