Synthesis, Cytotoxicity, Molecular Docking, Molecular Simulation and ADME Properties of Cinnamoylated Chloroquine Hybrid Analogues as Corona Virus Protease Inhibitors
K. R. Jayanthi, K. Hemapriya and Subban Ravi*
Department of Chemistry Karpagam Academy of higher Education, Coimbatore-21 India.
Corresponding Author E-mail: ravisubban@rediffmail.com
DOI : http://dx.doi.org/10.13005/ojc/380120
Article Received on : 20-10-2021
Article Accepted on :
Article Published : 07 Jan 2022
Reviewed by: Dr. Bashdar Ismael Meena
Second Review by: Dr. Haresh Thakellapalli
Final Approval by: Dr. Tawkir Sheikh
In an attempt to challenge COVID-19, molecular docking of cinnamoylated chloroquine compounds 1 – 15 against main protease (Mpro) enzyme of SARS-CoV-2 was undertaken. To study the stability of the complex formed between the drug and the receptor, suitable docking possesses were selected and put into molecular dynamics studies. Further ADME properties were determined using SWISS ADME software. In the docking studies compounds 5, 9, 14 and 15 exhibited encouragable binding with the Mpro crystal structure with docking scores of -8.1, -7.9, -7.8 and -7.9 Kcal/mole respectively. It was observed that CYS145 and GLU166 played a significant role during the interaction of molecules with the active site of COVID-19 Mpro. Among compounds 5, 9, 14 and 15, compound 5 had stable interactions with the protein, which might be the reason for the optimum RMSD, RMSF, radius of gyration and protein–ligand contacts (hydrogen bonding) values. The compound 5 was synthesised and tested for its cytotoxic activity against fibroblast L929 cell line. The above study indicated that the compound 5as a promising agent, and during the drug discovery process it could be taken as a starting point for lead optimization.
KEYWORDS:ADME properties; COVID-19; cytotoxic activity; Docking studies; SARS-CoV-2
Download this article as:| Copy the following to cite this article: Jayanthi K. R, Hemapriya K, Ravi S. Synthesis, Cytotoxicity, Molecular Docking, Molecular Simulation and ADME Properties of Cinnamoylated Chloroquine Hybrid Analogues as Corona Virus Protease Inhibitors. Orient J Chem 2022;38(1). |
| Copy the following to cite this URL: Jayanthi K. R, Hemapriya K, Ravi S. Synthesis, Cytotoxicity, Molecular Docking, Molecular Simulation and ADME Properties of Cinnamoylated Chloroquine Hybrid Analogues as Corona Virus Protease Inhibitors. Orient J Chem 2022;38(1). Available from: https://bit.ly/3JKj6p7 |
Introduction
Corona virus from the family Coronaviridae belongs to the order Nidovirales and are mostly found to be distributed in human beings 1. Once SARS-CoV-2 infects a cell, the host cell activities are totally controlled by the virus and its RNA gets translated to proteins with long chains, and in turn generate multiple copies. The viral proteases convert the long viral proteins into smaller pieces and activate them, and thereby play a critical role in the viral propagation. To treat the viral diseases like hepatitis C and HIV12 FDA has approved a good number of viral protease inhibitors (like darunavir, atazanavir, boceprevir, etc.) as drugs for viral diseases. The protease reported from SARS-CoV2 is a protein having the shape of an heart and comprises of dimers of two identical subunits. When the molecules bind to specific sites 3 the protease activity is triggered.
In an attempt to challenge COVID-19, an in silico study was made and in this regard virtual screening of 15cinnamoylated chloroquine hybrid analogues (1-15) against the crystal structure of the protease protein also known as 3CL hydrolase (Mpro) (PDB ID: 6Y2F) was carried out. This study aims to find an effective viral protease inhibitor and carried out the molecular docking study and also predicted ADME properties of these compounds 1-15. Among these molecules, compound 5 with the least score in molecular docking studies and with a favourable ADME property and later it was subjected to further studies like molecular dynamics simulation to investigate the stability of the complex formed. Further the compound was synthesised and subjected to in-vitro cytotoxic activity against fibroblast L929 cell line.
Materials and Method
Selection of drug target and Ligands
Cinnamoylated chloroquine hybrid analogues 1 – 15 were taken as the ligands and the protease protein with PDB ID: 6Y2F as the target protein for the in-silico studies. The X-ray crystal structure of the above protein complexed with a ∝ -ketoamide inhibitor was retrieved from the protein database.
Protocol for molecular docking studies
The Auto Dock 4.2 software package was used to carry out the docking. The ligands 1 – 15 were drawn and energy minimization of ligands were performed using a tool, known as Avogadro. Auto Dock and MGL tools were used for protein and ligand preparation and files which gives the output were saved in. pdbqt formats. After docking, the ligand–protein binding interaction was analysed using Discovery Studio 3.5, UCSF Chimera 1.13.1 and PyMOL 2.3. The binding energy was estimated with Mpro (PDB ID: 6Y2F) at resolution 1.95 Å. All the water molecules were removed in addition to the deletion of heteroatoms and co-crystal ligand from 6Y2F and then MGL tool was used to add Gasteiger charges and H- atoms and these are stored in. pdbqt format. The grid box dimension along x, y, and z axis was fixed at 40 × 40 × 40Å, with 0.375 Å spacing. All the input files, were made by Auto Dock graphical user interface. Lamarckian GA (4.2) and a genetic algorithm were used for docking simulation and evaluation. The Auto Dock tool was used to analyse the ligand – receptor interactions in their best binding poses and the lowest Gibbs free binding energy (estimated as ∆G in kcal/mol) conformers were selected for post-dock analysis.
Molecular dynamic simulations
The structure of the complex obtained from the protein 6Y2F and compound 5 was made ready for MD simulation. GROMACS-2021 version was employed to carry out 50 ns simulations using the CHARMM36 force field. For solvating the complexes, the SPCE water model was chosen followed by addition of ions to neutralize. Minimization of energy was performed with a tolerance of 1000 kJ/mol/nm. System equilibration was carried out for 50 ns using NVT and NPT ensemble. The NPT ensemble (T = 300 K and P = 1 bar) was carried out and the results of the protein-ligand complexes were analysed.
ADMET properties
Using the Swiss ADME software the various physicochemical and ADMET-related in silico properties were predicted.
Synthesis of compound 5
Preparation of compound (A)
4, 7-dichloroquinoline (100g) and 200 ml of ethylenediamine were taken in a RB flask and maintained at 110 ºC with continued stirring 24 hrs. Once the reaction is completed, the contents in the flask was transferred into a container with cold water; the content was continuously stirred for 120 min to yield a solid and then filtered. Then petroleum ether was used to wash the yielded solid and by means of a vacuum tray dryer the product was dried at 40 ºC (106 g, yield 97.4%).
Synthesis of compound B
The 4-chloro benzaldehyde (1.0g, 1.0eq) was taken in a 100.0 ml RB flask having 14.5 ml dimethylformide (DMF) and malonic acid (1.09 eq) and added with Diaza bicyclooctane (DABCO 1.1eq) which was used as a catalyst. The reaction mixture was heated to 110 0C and at the same temperature and retained for 2.0 hr. TLC was used to determine the Completion of the reaction. The reacted contents were poured into 50.0 ml water maintained at 5 ºC and using ethyl acetate 2 x 25ml the product was extracted. Using anhydrous Na2SO4 the ethyl acetate layer was dried, concentrated and the yielded solid was thoroughly washed with petroleum ether (10.0 ml), and vacuum dried at 45 ºC to yield B (3-(4-chloro phenyl) acrylic acid).
Compound B
Colour: White solid; Yield: 68.4 %, IR (cm-1, KBr,) :3432, 3018, 1631, 1532, 1425,1221, 776; 1H NMR (Deuterated acetone, 400 MHz) δ :7.69 (2H, d,J = 8.1Hz), 7.56 (1H, d, J = 15.9Hz,), 7.37 (2H, d, J = 8.1Hz), 6.45 (1H, d, J = 15.9Hz); MS: ESI positive mode (m/z):182 [M+H]+ .
To compound B (1.0eq) was dissolved in 20 ml of THF and it was maintained at 0 0C, then added 1.2 eq of dicyclo hexyl dicarbodimide (DCC), stirred for 20 min. Then added 1.2eq of N-hydroxy benzo triazine (HOBT), stirred for 40 min and to this a solution of compound-A (1.2 eq) was added dropwise at 0 ºC for 80 min. The reaction mixture was kept overnight at 20-27ºC and monitored by TLC. Once the reaction gets completed the mass was poured in to 50 ml cold water and extracted with chloroform 3x30ml. The chloroform layer was dried, concentrated under vacuum, added with 20 ml THF and kept at 0 ºC for 24 hrs. The yielded solid was filtered, vacuum dried at 50 ºC for 4 hr to get compound 5
Compound 5: N-(2-(7-chloroquinolin-4-ylamino) ethyl)-3-(4-chlorophenyl) acrylamide
Colour: White solid; Yield: 55.0%, 1HNMR (CD3OD + CDCl3, 400MHz) δ:8.41 (1H, d, J =5.56), 7.87(1H, d, J =8.32), 7.67(1H, d, J =2.14), 7.44(1H, d, J =15.71), 7.39 (2H, d, J =8.32), 7.29(1H, dd, J =2.21, 7.89), 7.29(2H, d, J =8.38), 6.54-6.39(2H, m,), 3.69-3.65(2H, m,), 3.53-3.49(2H, m). MS: ESI positive mode (m/z) 386.4[M+H]+ and Anal. Calcd. for C20H17Cl2N3O: C,62.18; H, 4.43; N, 10.87; Found: C,62.19; H, 4.45; N, 10.83.
In-vitro cytotoxic activity of compound 5
From National Centre for Cell Sciences, Pune, India, the Fibroblast L929 cell was procured and was cultured in a dedicated flask with Dulbecco’s modified Eagles medium. It was added with 10% FBS, sodium bicarbonate, L-glutamine and antibiotic solutions as supplements.
This was kept at 37ºC in an incubator (NBS Eppendorf, Germany) humidified with 5% CO2. Inverted phase contrast microscope was used to evaluate the viability of cells. MTT assay was used to evaluate the cytotoxic activity.
Cytotoxicity Evaluation:
The confluent cells containing approximately 5×103 cells/well was suspended in 96 well plates and allowed to grow in the incubator for 24 hrs. Then the growth medium was removed and 100µl of fresh solution of compound 5 in DMEM in various concentration (100µg, 50µg, 25µg, 12.5µg, 6.25µg in 500µl of DMEM) were added and allowed to grow in the same condition. The cells not treated with any one the compounds act as control and was maintained in the same environment.
Cytotoxicity Assay by Direct Microscopic observation:
After one full day of treatment the plate was investigated through a microscope (Olympus CKX41) and the images were recorded. If any remarkable changes are detected with respect to the cell morphology, like vacuolization of cells, rounding of cells, shrinking of cells, and presence of granulation in the cytoplasm of the cells then they cytotoxicity is indicated.
Cytotoxicity by MTT Assay:
After 24 hours, from the wells the sample content was removed and MTT solution of about 30µl was added, well mixed and kept for another 240 min. at 37ºC in the same incubator (Laura B. Talarico et al., 2004). After that, the supernatant was removed and the resulted formazan crystals were dissolved in 100µl of DMSO (Sigma Aldrich, USA) and absorbance values were measured at 540 nm. The viability percentage was determined using the formula, the ratio between the mean OD of the sample x 100 to the mean OD of control. The observed results are exhibited in the table 1. For the compound 5 the LC50 value was determined as 214.092 µg/mL. This shows that the compound was not cytotoxic in nature.
Table 1: The optical density values obtained for the compound 5 by MTT assay.
|
Sample Concentration (µg/mL) |
OD value I |
OD value II |
OD value III |
Average OD |
Percentage Viability |
|
6.25 |
0.6731 |
0.6727 |
0.6763 |
0.6740 |
98.54 |
|
12.5 |
0.6325 |
0.6315 |
0.6328 |
0.6323 |
92.44 |
|
25 |
0.6156 |
0.6194 |
0.6117 |
0.6156 |
90.00 |
|
50 |
0.5531 |
0.5555 |
0.5634 |
0.5573 |
81.48 |
|
100 |
0.5221 |
0.5285 |
0.5227 |
0.5244 |
76.67 |
|
Control |
0.6864 |
0.6839 |
0.6817 |
0.6840 |
100.00 |
Results and Discussion
It is widely discussed in the literature that chloroquine (CQ) and hydroxychloroquine (HCQ) are FDA approved drugs for malaria and autoimmune diseases and they increase the endosomal pH to inhibit viral infections 4-7. Earlier in our laboratory a series of novel hybrid molecules of cinnamoylated chloroquines were evaluated for malaria 8. From this series15 compounds (Fig 1) 1 – 15 were taken for the present study. One of our objective of taking up of this work with these selected molecules 1-15 is to find out where these molecules bind inside the Mpro binding site and in turn it might help us to consider them as a probable template which can be used further for the development of new Mpro viral infection inhibitors.
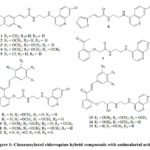 |
Figure 1: Cinnamoylated chloroquine hybrid compounds with antimalarial activity |
The molecular docking and binding energy estimation were carried out for the above 15 compounds with the documented structure of the viral protein Mpro (PDB ID: 6Y2F) at 1.95 Å resolution. With a positive control the optimised docking protocol was validated. The co-crystal ligand was docked once again into the catalytic binding site of 6Y2F using the Auto Dock tool. Also using the PyMol application the RMSD value was determined between the docking and co-crystal ligand poses. The docking protocol was validated by obtaining a low RMSD value (<1.2). Conformers with lowest energy has been carried forward for the post-docking analysis The results were presented in the Table-2.
Table 2: Docking scores (Kcal/mole) for the compounds 1-15 using Auto dock
|
Compound |
Binding Score |
Compound |
Binding Score |
|
1 |
-7.5 |
9 |
-7.9 |
|
2 |
-7.3 |
10 |
-6.7 |
|
3 |
-7.2 |
11 |
-6.9 |
|
4 |
-7.3 |
12 |
-6.6 |
|
5 |
-8.1 |
13 |
-7.3 |
|
6 |
-6.8 |
14 |
-7.8 |
|
7 |
-6.5 |
15 |
-7.9 |
|
8 |
-7.5 |
The 6Y2F structure consists of two protomers identical with each other and it is a homodimer composite. There are three domains in each monomer. Domain I is made up of the amino acid residues 8 – 101, and this was followed by domain II which is made up of residues 102 – 184. The domain III is in the form of a globular cluster and consists of five α-helices and this arrangement controls the protein dimerization. A cleft and a chymotrypsin fold are formed in between the domain I and II and they act as a substrate binding pocket. A long-extended loop connects domain II and III and consists of residues 185 – 200. In addition to this an extra domain is also present which consists of residues in the C-terminal. The active site amino acid residues which are present in the I and II domains are further divided in to subsites S1–S6, and these sites act as substrate binding pockets for the inhibitors.
In the S1 subsite the residues His41–Cys145 collectively called as the catalytic dyad is present and the subsites S2 and S4 consists of hydrophobic side chains. During proteolysis in order to stabilise the transition process the residues CYS145, SER144, and GLY143 interact with a conserved GLN carboxylate anion present in the cleavage site and forms an oxyanion hole which is an essential function of the S1 subsite [9,10]. In the present study, the binding mode analysis of compounds 1 – 15 with Mpro exhibited favourable hydrogen bonding interactions along with alkyl and mixed π-alkyl hydrophobic interactions at the active binding pocket, as shown in Fig. 2 – 5. Among the compounds docked compound 5, 9, 14 and 15 showed good scores and interactions. Rather than one Among the compounds docked compound 5, 9, 14 and 15 showed good scores and interaction. Rather than one hydrogen-bonding interaction, it is always advantageous to have multiple hydrogen-bonded interactions with the catalytic centre (S1 subsite) of the target proteases 9,10.
The amine hydrogen of the compound 5 (fig 2) showed a tri-center H- bond interaction to the oxygen of His A: 164 (2.87 A) and Cys A: 145 (2.27 A) and was found to be present firmly and deeply in the S1 pocket of the protease. The NH hydrogen of ARG A: 188 formed an H- bond with the amide carbonyl oxygen of 5. Further the π electrons of the quinolone moiety and the chloro substituted benzene ring showed hydrophobic Pi-alkyl interaction with CYS A:145 and PRO A:168 respectively. Van der Waals interactions were observed with MET A 165, GLU A:166, HIS A: 41, MET A:49, GLY A: 143 and THR A: 190.
 |
Figure 2: Interactions of compound 5with the protein PDB ID: 6Y2F |
Similarly compound 9 (fig 3) was also found to be seated in the S1 pocket of the protease and nitrogen atom of the quinolone moiety formed a hydrogen bond interaction to the oxygen of Cys A: 145 (2.86 A) and. Further the amide NH formed an hydrogen bond with C=O of GLU A: 166 (2.29 A). Further the π electrons of the aromatic ring showed hydrophobic Pi-alkyl interaction with MET A 165 and PRO A:168. Van der Waals interactions were observed with, MET A:49, HIS A:169, GLN A: 142 and THR A: 190.
 |
Figure 3: Interactions of compound 9with the protein PDB ID: 6Y2F |
In the compound 14 (fig 4) the hydroxy hydrogen formed a tri-center hydrogen bonded interaction to the oxygen of Cys A: 145 (2.29 A) and SER A: 144 (2.87 A) and LEU A:141(2.96 A). Further the π electrons of the quinoline moiety and they showed hydrophobic Pi-alkyl interaction with HIS A:41 and MET A:165. Van der Waals interactions were observed with GLU A:166, HIS A:164, ARG A:188, GLY A: 143 and THR A: 190.
 |
Figure 4: Interactions of compound 14with the protein PDB ID: 6Y2F |
More interestingly in the compound 15 (fig 5) the hydroxy hydrogen exhibited tri-center hydrogen bond interaction to the oxygen of Cys A: 145 (2.27 A), ASN A: 142 (2.75 A), SER A:144 (2.60 A) and GLY A 143(2.87 A). Further the π electrons of the quinoline moiety showed hydrophobic Pi-alkyl interaction with HIS A: 141, and MET A 165. Van der Waals interactions were observed with, GLU A:166, HIS A:164, MET A:49, ARG A: 188 and THR A: 190.
 |
Figure 5: Interactions of compound 15with the protein PDB ID: 6Y2F |
In compound 9, the amide NH formed an hydrogen bond with GLU A: 166 (2.29 A). It was reported that GLU 166 has a significant role in the dimerization of two protomers and also it helps in the shaping of S1 pocket of the substrate-binding site 49. Due to this reason compound 9 can effectively inhibit the functions of 6Y2F. With respect to other ligands, they showed H-bonded interaction either with CYS145 and/or HIS41 residues of the active pocket’s catalytic domain, which suggests that these two amino acids plays a critical role for inducing Mpro inhibitory activity.
Molecular dynamics (MD) simulations studies of compound 5 with 6Y2F
Based on the interactions made by the compounds 5, 9, 14 and 15 in the binding site and the respective binding energy determined (Fig. 2-5, Table 1), we have chosen the docked complex of compound 5 to run Molecular Dynamics (MD) simulation. Evaluation ws made based on the stability criteria during the MD simulation study using GROMACS 2021 package. To validate, confirm, and study the time-dependent interactions of ligand 5 with 6Y2F and the stability of the complex formed between the ligand and the receptor, simulation was carried out for 50 ns, and the dynamic stability of the complex was studied using the characteristics like protein–ligand RMSD, L-RMSF, radius of gyration and protein–ligand contacts (hydrogen bonding). The study revealed that during the simulation period the energy value of compound 5 was consistent and indicates that the Compound 5 possess the required stable structure for the drug designing processes.
Analysis of root mean square deviations of protein (RMSD)
RMSD analysis was carriedout to investigate the conformational stability of the ligand-protein complex formed between compound 5 and the protein 6Y2F and to study the conformational stability of the protein 6Y2F backbone. It is investigated to find out whether there is any movement of atoms in the protein 6Y2F when it is bound with the compound 5 in its active sites. RMSD plot score was calculated for 50 ns. The average backbone RMSD for unbound and bound complex (between compound 5 and 6Y2F) varied between 0.1–0.22 nm and found to be stable for the entire 50 ns for which MD simulation was carried out.
 |
Figure 6: RMSD of solvated SARS-CoV-2 M pro protein backbone and in complex with ligand 5 during 50 ns molecular dynamics simulation |
Protein root mean square fluctuation (RMSF).
The structural integrity of both Mpro ligand complex and the protein backbone were predicted by calculating the RMSF value. This also helps to determine the binding stability of the protein with the ligand throughout the period of simulation. The results of the RMSF calculations made for the MPro ligand complex and for the protein were presented in the form of a graph in Fig.7. Each and every peak in this graph, represents the protein area that fluctuates to a larger extent during the period of simulation. Usually the N-terminal and C-terminals, the so called ‘tails’ fluctuate to a greater extent when compared with other residues present in the protein. The average RMSF value obtained for the protein and the protein complex formed between the protein and the compound 5 was 0.15 nm. The result suggested that compound 5 binding with the protein 6Y2F substantially decreased the RMSF values.
 |
|
Protein–ligand contacts
The investigation of interactions taking place between the protein and the ligand (compound 5) more important as it gives the information about the specific target sites. During molecular docking analysis of compound 5 it (fig 2) was firmly placed inside the S1 pocket of 6Y2F protein and amine hydrogen exhibited hydrogen bonds with the oxygen of Cys A: 145 (2.27 A) and His A: 164 (2.87 A). Further the amide carbonyl oxygen formed an hydrogen bond with NH hydrogen of ARG A: 188 (3.24 A). In the MD simulation study, the interactions existed between the compound 5 and 6Y2F were found stable over the simulation period (Fig 8,9).
 |
|
 |
Figure 9: Plot of hydrogen bond distribution within the SARS-CoV-2 M pro protein, SARS-CoV-2 Mpro protein complex with ligand 5 |
Radius of gyration (Rg)
Radius of gyration (Rg) is a, important parameter used to assess the folding of a protein from its regular secondary structure into its three dimensional structure. Rg is used not only to identify any change is there in protein structure compactness but also gives information about its overall dimension. During binding of compound 5, initially (upto 30 ns) the Rg value was 2.15 nm for both ligand bound and unbound protein and latter from 30 ns and 50 ns of simulation the value was 2.2. This suggests that the secondary structure of the protein was not disturbed during binding of the compound 5 throughout the simulation period. The result strongly indicate that tight packing of the protein was observed even after binding of the ligand and thus making a stable complex (Fig 10).
 |
Figure 10: Rg during 50 ns molecular dynamics simulation of SARS-CoV-2 M pro protein and in complex with ligand 5 |
In Silico ADME properties of selected ligands 5, 9, 14 and 15
Table 2 exhibits the results of the ADME characteristics (absorption, distribution, metabolism, and excretion) determined using Swiss ADME package. Through this study the physicochemical properties of the compounds, their biological functions and the drug likeness of a compound could be determined. These properties like solubility, ionization (pKa), permeability, lipophilicity, transporters, and pharmacokinetic parameters are used to predict the efficacy of the compounds 5, 9, 14 and 15. The results arepresented in Table-3. It was observed from the table with respect to the pharmacokinetics properties that compounds 5, 9, 14 and 15 qualifies the drug likeness criteria. No violations of Lipinski’s rule of five was observed.
Below table shows the ADME properties of compounds 5, 9, 14 and 15 determined using Swiss ADME
Table 3: (A) Physicochemical parameters of compounds 5, 9, 14 and 15
|
Molecule |
Vina Score Kcal/mol |
MW |
No. Heavy atoms |
No. Rotatable bonds |
No. H-bond acceptors |
No. H-bond donors |
MR |
TPSA |
|
5 |
-7.4 |
386.27 |
26 |
7 |
2 |
2 |
108.1 |
54.02 |
|
9 |
-7.9 |
546.01 |
39 |
13 |
6 |
2 |
152.28 |
98.78 |
|
14 |
-6.6 |
562.06 |
40 |
14 |
7 |
3 |
158.05 |
101.94 |
|
15 |
-7.4 |
532.03 |
38 |
13 |
6 |
3 |
151.56 |
92.71 |
Table 3: (B) Solubility parameters of compounds 5, 9, 14 and 15
|
Molecule |
iLOGP |
iLOGP |
XLOGP3 |
WLOGP |
MLOGP |
Ali Log S |
Bioavailability Score |
|
5 |
3.26 |
3.26 |
5.44 |
4.48 |
3.31 |
-6.33 |
0.55 |
|
9 |
4.1 |
4.1 |
5.53 |
5.11 |
2.15 |
-7.36 |
0.55 |
|
14 |
4.95 |
4.95 |
5.13 |
4.94 |
2 |
-7.02 |
0.55 |
|
15 |
4.42 |
4.42 |
5.16 |
4.93 |
2.34 |
-6.85 |
0.55 |
Table 3: (C) Pharmacokinetic parameters of compounds 5, 9, 14 and 15
|
Molecule |
GI absorption |
BBB permeant |
Pgp substrate |
CYP1A2 inhibitor |
CYP2C19 inhibitor |
CYP2C9 inhibitor |
CYP2D6 inhibitor |
|
5 |
High |
Yes |
No |
Yes |
Yes |
Yes |
Yes |
|
9 |
High |
No |
Yes |
No |
Yes |
Yes |
Yes |
|
14 |
High |
No |
Yes |
Yes |
Yes |
Yes |
No |
|
15 |
High |
No |
Yes |
Yes |
Yes |
Yes |
Yes |
Table 3: (D) Drug likeliness rules of compounds 5, 9, 14 and 15
|
Molecule |
Lipinski rule, No. of violations |
Ghose rule, No. violations |
Veber rule, No. of violations |
Egan rule No. of violations |
Muegge rule, No. of violations |
|
5 |
0 |
0 |
0 |
0 |
1 |
|
9 |
1 |
2 |
1 |
0 |
1 |
|
14 |
1 |
3 |
1 |
0 |
1 |
|
15 |
1 |
2 |
1 |
0 |
1 |
Synthesis of compound 5
4, 7-dichloroquinoline reacted with ethylenediamine at 110 ºC under stirring for 24 hrs to yield .an intermediate compound A. Also 4-chloro benzaldehyde reacted with malonic acid in presence of a catalyst Diaza bicyclooctane (DABCO) to yield another intermediate B. Compound A and B react with each other in presence of dicyclohexyl dicarbodimide (DCC) to form the compound 5 (Scheme 1).
 |
Scheme 1: Synthesis of compound 5 |
Cytotoxic activity of compound 5
The concept behind the in vitro cytotoxicity test is that toxic compounds could affect the primary functions of cells which are present in common in all the tissues leading to determinable cellular structural and functional damage. The fibroblast cells are highly proliferative and are present in all types of connective tissues. In cytotoxicity evaluation with respect to cellular viability and proliferation, normally fibroblast cells are used. In this aspect, cytotoxicity testing with the cultures having fibroblast cell is considered as general bioassay, which provides appropriate information regarding basal cytotoxicity [11]. The cytotoxicity of the compound 5 was determined by MTT assay against fibroblast L929 cell lines. It behaved in a dose- and time-dependent manner. The LC50 value of the compound was calculated and the value obtained for the compound 5 is 214.092µg/mL. This shows that the compound was not cytotoxic in nature. The above data was complemented by the in-silico study and indicated that the compound 5 is a promising agent, and might act as a starting point for lead optimization in drug discovery.
Conclusion
The main protease (Mpro) enzyme of SARS-CoV-2 is an attractive drug target for antiviral drug design. In this aspect, molecular docking studies, molecular dynamics studies and predicted ADME properties was carried out for compounds 1-15. In the docking studies compounds 5, 9, 14 and 15 exhibited encouraging results the crystal structure of Mpro over the other selected molecules in this study with docking scores of -8.1, -7.9, -7.8 and -7.9 Kcal/mole respectively. The molecules interacted with CYS145 and GLU166 amino acids present in the active site of COVID-19 Mpro. Among compounds 5, 9, 14 and 15, compound 5 showed stable interactions with the protein and gave optimum RMSD, RMSF, radius of gyration and protein–ligand contacts (hydrogen bonding) values. Most of the analogs from 1-15 showed acceptable ADMET properties, with respect to GI absorption, P-gp interaction, and low toxicity. The compound 5 was synthesised and tested for its cytotoxic activity against fibroblast L929 cell line which showed a LC50 value of 214.092µg/mL. The above data suggested that the compound 5 is a promising agent, that might act as a beginning point for optimization in drug discovery.
Acknowledgment
The authors are thankful to Karpagam Academy of Higher Education Central Instrumentation Facility for providing facilities to characterise the compounds.
Conflict of Interest
The authors have no conflict of interest.
Funding Sources
There are no funding source.
References
- Richman, D. D.; Whitley, R. J.; Hayden, F. G. Clinical Virology. 2016, 4, 978-1-683-67316-3.
CrossRef - Himanshu, R.; Atanu, B.; Yash P. S.; Akhil, S.; Lovejit, S.; Gourav, S.; Usha, Y. N.; Vikash, K. D.; Gyan, M. Mol Divers, 2021, 25, 1905–1927.
CrossRef - Choudhary, M. I.; Shaikh, M.; Wahab, A.; Rahman, A.; Agency for Healthcare Research and Quality. 2020, 15(7), 0235030.
CrossRef - Devaux, C. A.; Rolain, J. M.; Colson, P.; Raoult, D. Int J Antimicrobl Agents. 2020, 55(5), 105938.
CrossRef - Touret, F.; Lamballerie, X. Antiviral Res, 2020, 177, 104762.
CrossRef - Wang, X.; Cao, R.; Zhang, H.; Liu, J.; Xu, M.; Hu, H.; Li, Y.; Zhao, L.; Li, W.; Sun, X.; Yang, X.; Shi, Z.; Deng, F.; Hu, Z.; Zhong, W.; Wang, M.; Cell Discovery. 2020, 6(28), 1-5.
CrossRef - Alim, A. B. Pharmacol. res. perspect. 2017, 5 (1), 1-13.
- Gayam, V.; Ravi, S. Eur. J. Med. Chem. 2017, 135, 382-391.
CrossRef - Marwa, A. A.; Fayed, E.; Inas, A.; Abdallah, M.; Hanan, E.; Ahmed, M.; Yassmin, M.; Khaled, A. M.; Abouzid, Y.; Elshaier, A. M. M.; Arab. J. Chem. 2021, 14, 103092.
CrossRef - Seketoulie, K.; Swapnil, P.; Seung, J. C.; Sci. Rep. 2020, 10, 17716.
- Freshney, R. I. J. Chem. Technol. Biotechnol. 2000, 45(4), 330.

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()



{kind=link}