Synthesis and Anti-Colon Cancer Activity of 1,2,4-Triazole Derivatives with Aliphatic S-Substituents
Sadiq Al-Mansury1, Asim A. Balakit*2 , Fatin Fadhel Alkazazz3, Kaiser N. Madlum4 and Rana A. Ghaleb4
, Fatin Fadhel Alkazazz3, Kaiser N. Madlum4 and Rana A. Ghaleb4
1Department of Pharmacology and Biochemistry, Veterinary Medicine College, Al-Qasim Green University, Babylon, Iraq.
2College of Pharmacy, University of Babylon, Babylon, Iraq.
3Department of Chemistry, College of Science, Al-Mustansiyriah University, Baghdad, Iraq.
4Department of Human Anatomy, College of Medicine, University of Babylon, Babylon, Iraq.
Corresponding Author E-mail: asim_alsalehi@hotmail.com
DOI : http://dx.doi.org/10.13005/ojc/350109
Article Received on : 16-11-2018
Article Accepted on : 27-01-2019
Article Published : 20 Feb 2019
A series of new 3-mercapto-1,2,4-triazoles have been designed, synthesized and their structures were identified by nuclear magnetic resonance (NMR) and Fourier transform infrared (FT-IR) spectrophotometric techniques. The target compounds are designed as analogues for the anti-cancer agent Combretastatin A-4 with different aliphatic side substituents. The synthesized novel heterocyclic compounds were evaluated as anticancer molecules against colon cancer cell line (SW480) using the crystal violet cytotoxicity assay. The results revealed that these compounds have growth inhibitive effect on the cancer cells with different inhibition levels. Compound 5a with -SMe group was found to be the most active one with 77.4% cell growth inhibition and 10 µM IC50 value, it was also found to have relatively low cytotoxicity when tested against Madin-Darby Canine Kidney (MDCK) normal cells line. The levels of the antioxidant total capacity of the synthesized triazoles have been determined by using 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay against ascorbic acid as a reference antioxidant agent at 50 μM. Compound 4 showed highest antioxidant activity with DPPH radical scavenging capacity of 71%.
KEYWORDS:Anticancer; Antioxidants; Colon Cancer; SW480; Triazole
Download this article as:| Copy the following to cite this article: Al-Mansury S, Balakit A. A, Alkazazz F. F, Madlum K. N, Ghaleb R. A. Synthesis and Anti-Colon Cancer Activity of 1,2,4-Triazole Derivatives with Aliphatic S-Substituents. Orient J Chem 2019;35(1). |
| Copy the following to cite this URL: Al-Mansury S, Balakit A. A, Alkazazz F. F, Madlum K. N, Ghaleb R. A. Synthesis and Anti-Colon Cancer Activity of 1,2,4-Triazole Derivatives with Aliphatic S-Substituents. Orient J Chem 2019;35(1). Available from: https://bit.ly/2BHS5RO |
Introduction
Cancer is considered as the second lethal disease worldwide,1 colorectal cancer is the third most common type of cancer around the world and it is one of the major causes of deaths related to cancer.2 The design and synthesis of anticancer agents is an interesting research area in which scientists are endeavoring to develop synthetic or natural compounds that can contribute in the treatment of cancer and help patients to be cured of this life-threatening disease.3 There is a very broad spectrum of natural compounds which are considered as anticancer agents4 among them combretastatin A‑4 (CA-4) which is natural product extracted and isolated from a South African plant; Combretum caffrum Kuntze (Combretaceae).5 The anticancer activity of CA-4 is attributed to its antitubulin polymerization activity by binding to the colchicine binding site.6 The simplicity of the structure of CA-4 and its remarkable anticancer efficacy make its structure a fundamental part of wide range of synthetic anticancer agents which are considered as combretastatin A-4 analogues.7-9 According to the structure activity relationship (SAR) investigations, it is reported the CA-4 analogues must have fundamental structural requirements for the optimal activity, the molecule must have 3,4,5-trimethoxyphenyl and 4-methoxyphenyl groups attached to two adjacent atoms (bridge), the two groups must be in cis-olefin configuration.10,11
A great attention has been devoted to the 1,2,4-triazol derivatives because of their biological activities, particularly sulfur-containing ones found to have diverse biological activities such as herbicidal,12 anticancer,13 antidepressant,14 antimicrobial,15 antifungal,16 analgesic/anti-inflammatory,17 antiviral,18 and antioxidant.19 Many studies have shown that the pharmacological activities of 1,2,4-triazole and their derivatives due to existence of the1,2,4-triazole nucleus.20 Some of compounds with triazole ring are reported as potent anticancer CA-4 analogues.21 The presence of S-linkers in the structure improve some drug-like parameters such as decreasing lipophilicity, increasing water solubility, and offering good hydrogen bond acceptors.22
Antioxidants are compounds that have the ability to reduce the oxidative stress by inhibiting the oxidation process leading to preventing the cell death resulting from generated free radicals.23 The excessive release of free radicals is intimately related to cancer disease,24 thereby antioxidants can contribute in fighting this deadly disease. There is huge number of both natural and synthetic compounds that are investigated for their antioxidant capacity.25,26
In this paper we report the synthesis of newly designed 1,2,4-triazoles with aliphatic substituents attached to the sulfur group, the new compounds are designed to meet the structural requirements of the highest expected antitubilin (anticancer) activity of the analogues of CA-4 to be tested as anticancer agents against colon cancer cell line SW480. The antioxidant capacity of the synthesized compounds is also studied.
Material and Methods
General
Chemicals and reagents were purchased from commercial sources and used without any further purification. 1H (400 MHz) and 13C NMR (100 MHz) spectra were recorded in CDCl3 or DMSO-d6 on a Bruker Bio Spin 400 MHz spectrometer. Chemical shifts δ (ppm) are reported relative to tetramethylsilane (TMS). IR spectra were recorded on a Tensor II, Bruker-Optics FT-IR spectrophotometer as KBr discs. Melting points were measured using Stuart SMP10 apparatus. Column chromatography was carried out using silica gel 200 – 300 Mesh. 3,4,5 Trimethoxybenzohydrazide was synthesized according to literature procedure.27
Synthesis: 4-(4-Methoxyphenyl)-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazole-3-thiol
4-Methoxyphenyl isothiocyanate (1.65 gm,10 mmol) was added to 3,4,5-trimethoxybenzohydrazide (2.263 g, 10 mmol) in 30 mL of ethanol, and refluxed for 4 h; then, left to cool to room temperature. The formed solid was isolated by filtration, washed with ethanol and derided. Then the precipitate was dissolved in 20 mL of 2 N NaOH and heated (refluxed) for 4h. The mixture was left to cool to room temperature and then filtrated; the filtrate was acidified with 10% HCL. The obtained solid was filtrated off and then washed with water.
Light-yellow solid; yield, 81%; m.p. 253-255°C; FT-IR: 3091-2931 (C-H, aromatic), 2829 (C-H, aliphatic), 2754 (SH), 1606 (C=N), 1591 (C=C) 1H NMR (400 MHz, DMSO-d6) δ: 3.57 (s, 6H, 2 x OCH3), 3.65 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 6.61 (s, 2H, Aromatic), 7.07 (d, J = 8.8 Hz, 2H, Aromatic), 7.30 (d, J = 8.8 Hz, 2H Aromatic), 14.07 (br, 1H, SH). 13C NMR (100-MHz, DMSO-d6) d: 56.0, 56.2 (2C), 60.5, 106.28 (2C), 115.0 (2C), 121.3, 127.8, 130.5 (2C), 139.4, 150.7, 153.0 (2C), 160.1, 169.3.
General Procedure for the Synthesis of Target Compounds (5a-j)
To the solution of 4 (0.373 gm,1 mmol) in 20 ml of 50% aqueous 2-propanol, NaOH (0.04 g, 1.5 mmol) was added with stirring for 5 mints, then appropriate alkyl halide (1.1 mmol) was added to. The reaction mixture was refluxed for 8 h. TLC technique was used to monitor the reaction, after finished, the 2-propanol was evaporated (under reduced pressure) and the product was extracted by chloroform, solvent was distilled off to give the crude solid product which was washed with hexane and then purified by column chromatography. The melting points, eluents and Rf values of the compounds 4, 5a-j are shown in Table 1.
Table 1: Melting points, eluents and Rf values of 4, 5a-j.
|
Compound |
M. P. °C |
Eluent (n-hexane : ethyl acetate) |
Rf (n-hexane : ethyl acetate; 1.5 : 2.5) |
|
4 |
253-255 |
— |
0.54 |
|
5a |
102-103 |
1: 2 |
0.07 |
|
5b |
80-81 |
1: 2 |
0.14 |
|
5c |
104-106 |
1: 1.5 |
0.285 |
|
5d |
106-108 |
1: 1.5 |
0.357 |
|
5e |
94-96 |
1: 1 |
0.297 |
|
5f |
112-113 |
1: 1.5 |
0.214 |
|
5g |
93-95 |
1: 1.5 |
0.2 |
|
5h |
90-92 |
1: 1.5 |
0.33 |
|
5i |
80-82 |
1: 1 |
0.19 |
|
5j |
109-110 |
1: 2 |
0.13 |
4-(4-Methoxyphenyl)-3-(methylthio)-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazole (5a)
Yellow solid, yield, 84%; FT-IR: 3090-2932 (C-H, aromatic), 2829 (C-H, aliphatic), 1607 (C=N), 1585 (C=C) 1H NMR (400 MHz, CDCl3) δ: 2.7 (s, 3H, SCH3), 3.63 (s, 6H, 2 x OCH3), 3.82 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 6.69 (s, 2H, Aromatic), 7.08 (d, J = 8.9 Hz, 2H, Aromatic), 7.20 (d, J = 8.9 Hz, 2H, Aromatic). 13C NMR (100 MHz, CDCl3) δ: 14.63, 55.69, 55.93(2C), 60.8, 105.37 (2C), 115.18 (2C), 121.3, 126.9(2C), 128.7, 139.8, 153 (2C), 154.1, 154.8, 160.6.
3-(Ethylthio)-4-(4-methoxyphenyl)-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazole (5b)
Yellow solid, yield 82%; FT-IR: 3091-2931 (C-H, aromatic), 2838 (C-H, aliphatic), 1607 (C=N), 1587 (C=C); 1H NMR (400 MHz, CDCl3) δ: 1.43 (t, J = 7.4 Hz, 3H, SCH2CH3), 3.3 (q, J = 7.3 Hz, 2H, SCH2CH3), 3.64 (s, 6H, 2 x OCH3), 3.82 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 6.69 (s, 2H, Aromatic), 7.03 (d, J = 8.9 Hz, d, 2H, Aromatic), 7.21 (d, J = 8.8 Hz, 2H, Aromatic). 13C NMR (100 MHz, CDCl3) δ: 14.7, 26.8, 55.7, 55.9(2C), 60.88, 105.3 (2C), 115.9 (2C), 121.9, 127.0, 128.7(2C) 138.8, 144.5, 153.0 (2C), 154.6, 160.5.
3-(Butylthio)-4-(4-methoxyphenyl)-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazole (5c)
Yellow crystal, yield 78%; FT-IR: 3001 (C-H, aromatic), 2958 (C-H, aliphatic), 1608 (C=N), 1588 (C=C); 1H NMR (400 MHz, CDCl3) 0.91 (t, J = 7.3 Hz, 3H, SCH2CH2CH2 CH3), 1.43 (sext, J = 7.3 Hz, 2H, SCH2CH2 CH2CH3), 1.7 (p, J = 7.4 Hz, 2H, SCH2 CH2CH2 CH3), 3.2 (t, J = 7.3 Hz, 2H, SCH2 CH2CH2 CH3), 3.64 (s, 6H, 2 x OCH3), 3.83 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 6.69 (s, 2H, Aromatic), 7.08 (d, J = 8.8 Hz, 2H, Aromatic), 7.19 (d, J = 8.8 Hz, 2H, Aromatic). 13C NMR (100 MHz, CDCl3) δ: 13.5, 21.8, 31.35, 32.2, 55.6, 55.9 (2C), 60.2, 105.3 (2C), 115.0(2C), 121.9, 127.1, 128.8 (2C), 139.1, 153, 153.5(2C), 154.6, 160.5.
3-(Hexylthio)-4-(4-methoxyphenyl)-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazole (5d)
White solid, yield 87%; FT-IR: 3110 (C-H, aromatic), 2836 (C-H, aliphatic), 1608 (C=N), 1588 (C=C); 1H NMR (400 MHz, CDCl3) δ: 0.87 (t, J = 6.7 Hz,3H, S(CH2)5CH3), 1.28-1.33 (m, 2H, S(CH2)4CH2CH3), 1.42 (p, J = 7.2 Hz, 4H, S(CH2)2CH2CH2CH2CH3), 1.77 (p, J =7.5 Hz, 2H, SCH2CH2(CH2)3CH3), 3.26 (t, J = 7.6 Hz 2H, SCH2(CH2)4CH3), 3.64 (s, 6H, 2 x OCH3), 3.8 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 6.69 (s, 2H, Aromatic), 7.08 (d, J = 8.5 Hz, 2H, Aromatic), 7.2 (d, J = 8.9 Hz, 2H, Aromatic). 13C NMR (100 MHz, CDCl3) δ: 13.9, 22.5, 28.4, 29.3, 31.2, 32.2, 55.6, 55.9(2C), 60.8, 105.33(2C), 115.03(2C), 121.8, 127.1, 128.8 (2C), 139.1, 153 (2C), 153.5, 154.6, 160.5.
3-(Isobutylthio)-4-(4-methoxyphenyl)-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazole (5e)
White solid, yield 78%; FT-IR: 3003 (C-H, aromatic), 2867 (C-H, aliphatic), 1606 (C=N), 1588 (C=C); 1H NMR (400 MHz, CDCl3) δ: 1.01 (d, J = 6.6 Hz, 6H, SCH2CH(CH3)2), 2.08-1.98 (m, 1H, SCH2CH(CH3)2), 3.18 (d, J = 6.8 Hz, 2H, SCH2CH(CH3)2), 3.6 (6H, 2 x OCH3), 3.83 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 6.69 (s, 2H Aromatic), 7.08 (d, J = 8.7 Hz, 2H, Aromatic), 7.19 (d, J = 8.7 Hz, 2H, ArH B). 13C NMR (100 MHz, CDCl3) δ: 21.8 (2C), 26.45, 40.9, 55.68, 55.89 (2C), 60.87, 105.3(2C), 115.03 (2C), 121.9, 127.1, 128.8 (2C), 139.1, 153.0 (2C), 153.1, 154.58, 160.5.
3-(Allylthio)-4-(4-methoxyphenyl)-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazole (5f)
Yellow solid, yield 87%; FT-IR: 3091 (C-H, aromatic), 2838 (C-H, aliphatic), 1607 (C=N), 1585 (C=C); 1H NMR (400 MHz, CDCl3) δ: 3.63 (s, 6H, 2 x OCH3), 3.82 (s, 3H, OCH3), 3. 85 (s, 3H, OCH3), 3.87 (d, J=7.1 Hz, 2H, SCH2CH=CH2), 5.12 (d, J=9.97 Hz, 1H, SCH2CH=CH2), 5.23 (d, J=9.2 1H, SCH2CH=CH2), 6.02-5.92 (m, 1H, SCH2CH=CH2), 6.69 (s, 2H, Aromatic), 7.08 (d, J = 8.8 Hz, 2H, Aromatic), 7.19 (d, J = 8.7 Hz, 2H, Aromatic). 13C NMR (100 MHz, CDCl3) δ: 35.38, 55.7, 55.9 (2C), 60.89, 105.33(2C), 115.03(2C), 119.0, 121.9, 127.1, 128.8 (2C), 132.65, 139.1, 152.6, 153 (2C), 154.7, 160.5.
3-(Isopropylthio)-4-(4-methoxyphenyl)-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazole (5g)
White solid, yield 83%; FT-IR: 2999 (C-H, aromatic), 2961 (C-H, aliphatic), 1607 (C=N), 1586 (C=C); 1H NMR (400 MHz, CDCl3) δ: 1.4 (d, J = 6.7 Hz, 6H, CH(CH3)2), 3.6 (s, 6H, 2 x OCH3), 3.82 (s, 3H, OCH3), 3. 86 (s, 3H, OCH3), 3.99-3.90 (m, 1H, CH(CH3)2, 6.69 (s, 2H, Aromatic), 7.0 (d, J = 8.9 Hz, 2H, Aromatic), 7.16 (d, J = 8.8 Hz, 2H, Aromatic). 13C NMR (100 MHz, CDCl3) δ: 23.5 (2C), 38.6, 55.7, 55.9 (2C), 60.8, 105.3 (2C), 115.03 (2C), 121.9, 127.2, 128.9 (2C), 139.1, 152.3, 153 (2C), 154.4, 160.4.
3-(Cyclohexylthio)-4-(4-methoxyphenyl)-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazole (5h)
White solid, yield 81%; FT-IR: 2936 (C-H, aromatic), 2848 (C-H, aliphatic), 1606 (C=N), 1589 (C=C); 1H NMR (400 MHz, CDCl3) δ: 1.2- 2.13 (m, 11H, cyclohexyl), 3.63 (s, 6H, 2 x OCH3), 3.81 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 6.68 (s, 2H, Aromatic), 7.0 (d, J = 7.7 Hz, 2H, Aromatic), 7.17 (d, J = 7.9 Hz, 2H, Aromatic). 13C NMR (100 MHz, CDCl3) δ: 25.57, 25.8, 33.4, 33.47, 46.48, 55.68, 55.8, 55.9 (2C), 60.89, 105.33(2C), 115.0 (2C), 121.9, 127.1, 128.8 (2C), 139.1, 152.6, 152.9 (2C), 154.4, 160.4.
4-(4-Methoxyphenyl)-3-((oxiran-2-ylmethyl)thio)-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazole (5i)
Light yellow solid, yield 79%; FT-IR: 2936 (C-H, aromatic), 2844 (C-H, aliphatic), 1601 (C=N), 1585 (C=C); 1H NMR (400 MHz, DMSO-d6) δ: 3.57 (s, 6H, 2 x OCH3), 3.65 (s, 3H, OCH3), 3. 79 (s, 3H, OCH3), 3.8 (d, J = 6.5 Hz, 2H, CH2 ring), 3.99-3.91 ( m, 1H, CH ring), 4.2-4.1 (m, 2H, SCH2), 6.57 (s, 2H, Aromatic), 7.08 (d, J = 8.8 Hz, 2H, Aromatic), 7.29 (d, J = 8.77 Hz, 2H, Aromatic). 13C NMR (100 MHz, DMSO-d6) δ: 48.03, 49.07, 56.03, 56.18, 60.6, 68.8, 105.6 (2C), 115.13 (2C), 122.5, 127.1, 129.9 (2C), 139.2, 144.35, 153.1(2C), 153.8, 159.89.
Methyl2-((4-(4-methoxyphenyl)-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazol-3-yl)thio)acetate (5j)
Brown solid, yield 83%, FT-IR: 3069-2935 (C-H, aromatic), 2961 (C-H, aliphatic), 1735 (-C=O), 1610 (C=N), 1587 (C=C); 1H NMR (400 MHz, CDCl3) δ: 3.6 (s, 6H, 2 x OCH3), 3.77 (s, 3H, OCH3, ester), 3.82 (s, 3H, OCH3), 3. 86 (s, 3H, OCH3), 4.15 (s, 2H,-SCH2COOCH3) , 6.69 (s, 2H, Aromatic), 7.02 (d, J = 6.9 Hz, 2H, Aromatic), 7.22 (d, J = 7.2 Hz, 2H, Aromatic); 13C NMR (100 MHz, CDCl3) δ: 34.2, 52.9, 55.7, 56.9 (2C), 60.9, 105.32 (2C), 115.22 (2C), 121.5, 126.6, 128.7 (2C), 139.2, 151.4, 153.8 (2C), 154.9, 160.7, 168.8 (C=O).
Culturing of SW480 and MDCK Cell Lines
The stock human colon cancer (SW480) and Madin-Darby Canine Kidney (MDCK) cell lines were obtained from the Cancer Research Lab. and Cell Culture at the College of Medicine, University of Babylon. The cells were grown as monolayer and maintained as an exponentially growth phase in Dulbecco’s modified Eagle’s medium (DMEM) with 1% penicillin-streptomycin (GIBCO®) and 5% heat inactivated fetal bovine serum (FBS) in tissue culture flask and incubated at 37ºC in humid enviroment containing CO2 (5%). The cells were preserved by the replacement of fresh medium.
Anticancer Activity
A 10 mM solutions of the triazoles (4, 5a-j) in acetone as solvent were prepared and diluted with DMEM to get the desired concentrations for the treatments of cell lines. Mixture of acetone: DMEM (200 μl, with the same ratios used in the test samples) was added to the negative control samples, the added volumes of acetone have no any effect on the cell growth when compared with the positive control samples, accordingly a normalized control was depended in the study.
Overnight cultures of the cells grown in 25 cm2 tissue culture flask were examined under inverted microscope. Cells were detached using trypsin, washed twice by adding 5 ml of PBS and centrifugation at 1500 rpm for 5 minutes. These exponentially growing cells were seeded in plates 96-well, 5000 cells /well and incubated at 37°C overnight (24 h). Then, the media was removed and 200 μl of fresh medium including the different concentrations of the test compounds (4, 5a-j) were added in four replicates and incubated at 37°C for 48 h. The wells were then washed by adding 200 μl of PBS for each well, left for 5 minutes, and removed by sterile pasture pipet. This process was repeated twice. The wells were treated with 100 μl of crystal violate solution for 10 minutes. The dye was discarded and the plates flushed three times with distilled water. The plates were left to dry at room temperature. The absorbance was recorded at 570 nm. The growth inhibition percentage was determined according to the below equation: 3,27
% inhibition = [(Abs control – Abs samples) / (Abs control)]* 100
Determination of IC50 Value
The IC50 of 5a was determined by treating the cancer cell line (SW480) with different concentrations of 5a (5, 10, 20, 40, 50 and 60 µM) to find out the concentration that results a 50% cell growth inhibition. The IC50 value was calculated by using SigmaPlot version 12 software, Figure 1.
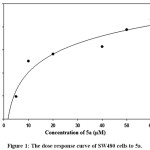 |
Figure 1: The dose response curve of SW480 cells to 5a. |
Antioxidant Activity
The DPPH radical scavenging protocol was applied to assess the scavenging activity of synthetized compounds. Solutions of the test compounds (0.005 M in DMSO) were prepared, then, 50 μM solutions were prepared by dilution with methanol. Ascorbic acid (50 μM) was also prepared in the same manner. In test tube, 3 mL of the test compound were mixed with 50 μL of DPPH (0.004 M in methanol), the mixtures were kept in dark place at 25°C for 30 min, ascorbic acid (AA) was used as standard antioxidant. The absorbance value was measured at 518 nm. The percentage of scavenging activity was determined according to the below formula:
%Scavenging of DPPH = [(Ac – As) / Ac] X 100
Where: Ac is the absorbance of the control (no sample or standard added to the DPPH solution) and As is the absorbance of the test. The tests were done in triplicate.
Statistical Analysis
Data were processed using SigmaPlot version 12.0 software. Student’s t-test was applied to locate the statistical significance for unpaired observations. The P values less than 0.01 were treated as significant results.
Results and Discussion
Synthesis
The target compounds (4, 5a-j) have been synthesized via three steps synthetic pathway (Scheme 1). The starting materials have been chosen to get the structural requirements that make the new compounds as CA-4 analogues. The structure of compound 4 is the simplest in the series, it is considered as the starting material for the synthesis of compounds 5a-j, the modification of the structure is done by the attaching the aliphatic groups to the –S atom (-S linker). In the first step, 3,4,5‑trimethoxybenzohydrazide (1) was reacted with 4-methoxyphenyl isothiocyanate (2) to give the intermediate (3) which was isolated and then treated with NaOH to achieve the intramolecular cyclization giving the 1,2,4-triazole derivative 4. In the last step the hydrogen atom of the –SH group of compound 4 was substituted by different aliphatic substituents upon treatment with the appropriate alkyl halide in basic medium. Relatively good isolated yields (78 – 84 %) were obtained after purification by column chromatography.
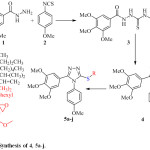 |
Scheme 1: Synthesis of 4, 5a-j. |
Anticancer Activity
To evaluate the anticancer activity of compound 4 and study the impact of structure modification on the activity of compounds 5a-j, human colon cancer cell lines (SW480) were treated with two different concentrations (10 µM and 50 µM) of compounds 4, 5a-j for a period of 48 h, the cell growth inhibition was determined using crystal violet cytotoxicity assay, Figure 2 shows the effect of the compound on the cells viability. The cell growth inhibition was calculated, Table 2 shows the obtained results, treatment of the cancer cell lines (SW480) with compound 4 which has free –SH group caused 6 and 10% cell growth inhibition at 10 µM and 50 µM respectively. Substitution of the H atom of the –SH group by methyl (compound 5a) caused a remarkable increment in the cytotoxic activity where the values of cell growth inhibition were found to be 50.4 and 77.4% at 10 µM and 50 µM respectively, on the other hand, the substitution of the H atom with primary aliphatic straight chains (n-ethyl, n-butyl and n-hexyl substituents in compounds 5b, 5c and 5d respectively) caused moderate improvement in the cytotoxic activity, within the results related to compounds 5a-d it is clear that increasing the chain length decreases the activity, while no big variation in the cytotoxicity levels was observed with the other compounds where branched, olefinic, cyclic or ester groups are present as S-substituents in the structures of compounds 5e-j.
The results indicated that 5a is the most potent cytotoxic agent in the series, accordingly the next step was to evaluate the IC50 of compound 5a against the same cell line, the results showed that 10 µM is the required concentration to cause 50% cancer cell lines (SW480) growth inhibition after incubation for 48 h. Furthermore, to test the cytotoxicity of 5a against normal cells, MDCK normal cell lines were treated with 5a at 10 µM concentration (IC50) for 48 h, only 20% cell growth inhibition was observed, see Figure 2 and Table 2.
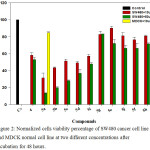 |
Figure 2: Normalized cells viability percentage of SW480 cancer cell line and MDCK normal cell line at two different concentrations after incubation for 48 hours. |
Table 2: Cytotoxicity levels of 4, 5a-j against cancer and normal cells at different concentrations for 48 h.
| Compound | Percentage of cell growth inhibition for SW480 | IC50( μM) | Percentage of cell growth inhibition for MDCK normal cells at IC50 | |
| 10 μM | 50 μM | |||
| 4 | 6 | 10 | — | — |
| 5a | 50.4 | 77.4 | 10 | 20 |
| 5b | 26.2 | 61.1 | — | — |
| 5c | 18 | 52.2 | — | — |
| 5d | 16 | 36.3 | — | — |
| 5e | 3 | 13 | — | — |
| 5f | 6.5 | 22 | — | — |
| 5g | 1 | 11 | — | — |
| 5h | 3 | 25 | — | — |
| 5i | 3.4 | 13 | — | — |
| 5j | 3.7 | 20 | — | — |
Antioxidant Activity
The synthesized compounds (4, 5a-j) were subjected to DPPH assay to determine their antioxidant activity. Table 3 include the obtained results, compound 4 exhibited the highest antioxidant activity (71%) when compared with ascorbic acid (80% as the reference antioxidant), this could be attributed to the availability of free –SH group that makes the compound capable to accommodate the free radical and stabilize it by delocalization. Compounds 5e, 5f, 5i and 5j showed low to moderate antioxidant activity while the others (5a-d, 5g and 5h) did not show any level of free radical scavenging activity.
Table 3: Results of DPPH assay for antioxidant activity.
| Compound | % Scavenging of DPPH free radical at 50 µM* |
| ascorbic acid (standard) | 80 |
| 4 | 71 |
| 5e | 24 |
| 5f | 10 |
| 5i | 45 |
| 5j | 18 |
Conclusion
Series of 3-mercapto-1,2,4-triazoles have been designed as CA-4 analogues with different aliphatic substituents attached to the S atom (4, 5a-j). The target compounds were synthesized and characterized by different spectroscopic techniques. The cytotoxicity of 4, 5a-j was evaluated against colon cancer line (SW480), compound 5a was found to be the most potent one and showed a kind of selectivity to cancer cells when subjected to evaluating its cytotoxicity against MDCK normal cell lines at 10 µM concentration (IC50). The results revealed 5a is a promising anticancer agent to be subjected for further studies. The antioxidant activity of 4, 5a-j was studied, the order of the activities was found to be 4 ˃ 5i ˃ 5e ˃ 5j ˃ 5f.
Acknowledgments
The authors thank Al-Qasim Green University, Al-Mustansiyriah University and the University of Babylon for funding the project.
References
- Jemal, A.; Bray, F.; Center, M. M.; Ferlay, J; Ward, E.; Forman, D.; CA Cancer J. Clin. 2011, 61, 69–90.
- Alhurry, A. M.; Rezaianzadeh, A.; Rahimikazerooni, S.; Akool, M. A.; Bahrami, F.; Shahidinia, S. S.; Pourahmad, M.; Annals of Colorectal Research 2017, 5, 3-4.
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G.; Nat. Chem. 2016, 8, 531-541.
- Cragg, G. M.; Grothaus, P. G.; Newman, D. J.; J. Nat. Prod. 2014, 77, 703-723.
- Pettit, G. R.; Singh, S. B.; Niven, M. L.; Hamel, E.; Schmidt, J. M.; J. Nat. Prod. 1987, 50, 119-131.
- Burns, R. G.; FEBS Lett. 1992, 297, 205-208.
- Kamal, A.; Bajee, S.; Nayak, V. L.; Rao, A. V.; Nagaraju B.; Reddy C. R.; Sopanrao, K. J.; Alarifi, A.; Bioorg. Med. Chem. Lett. 2016, 26, 2957-2964.
- Patel, V. K.; Harish, R.; Bioorg. Med. Chem. Lett. 2016, 26, 2115-2118.
- Engdahl, A. J.; Edith, A. T.; Sarah, E. L.; Taylor, B. E.; Pamela, S. M.; Craig, N. S.; Org. Lett. 2015 17, 4546-4549.
- Hsieh, H. P.; Liou, J. P.; Mahindroo, N.; Curr. Pharm. Des. 2005, 11, 1655-1677.
- Romagnoli, R.; Baraldi, P. G.; Prencipe, F.; Oliva, P.; Baraldi, S.; Tabrizi, M. A.; Lopez-Cara, L. C.; Ferla, S.; Brancale, A.; Hamel, E.; Ronca, R.; Sci. Rep. 2016, 6, 26602.
- Wang, B. L.; Shi, Y. X.; Ma, Y.; Liu, X. H.; Li, Y. H.; Song, H. B.; Li, B. J.; Li, Z. M.; J. Agric. Food Chem. 2010, 58, 5515-5522.
- Kamboj, V. K.; Verma, P. K.; Dhanda, A.; Ranjan, S.; Cent. Nerv. Syst. Agents Med. Chem. 2015, 15, 17-22.
- Bhatnagar, A.; Sharma, P. K.; Kumar, N.; Int. J. PharmTech. Res. 2011, 3, 268-282.
- Karabasanagouda, T.; Adhikari, A. V.;Shetty, N. S.; Eur. J. Med. Chem. 2007, 42, 521-529.
- Sztanke, K.; Tuzimski, T.; Rzymowska, J.; Pasternak, K.; Kandefer-Szerszeń M.; Eur. J. Med. Chem. 2008, 43, 404-419.
- Sarigol, D.; Uzgoren-Baran, A.; Tel, B. C.; Somuncuoglu, E. I.; Kazkayasi, I.; Ozadali-Sari, K.; Unsal-Tan, O.; Okay, G.; Ertan, M.; Tozkoparan, B.; Bioorg. & Med. Chem. 2015, 23, 2518-2528.
- Küçükgüzel, I.; Tatar, E.; Küçükgüzel, Ş. G.; Rollas, S.; De Clercq, E.; Eur. J. Med. Chem. 2008, 43, 381-392.
- El-Sherief, H. A.; Youssif, B. G.; Bukhari, S. N.; Abdel-Aziz, M.; Abdel-Rahman, H. M.; Bioorg. Chem. 2018, 76, 314-325.
- Zhang, Q.; Peng, Y.; Wang, X. I.; Keenan, S. M.; Arora, S.; Welsh, W. J.; J. Med. Chem. 2007, 50, 749-754.
- Blanch, N. M.; Chabot, G. G.; Quentin, L.; Scherman, D.; Bourg, S.; Dauzonne, D.; Eur. J. Med. Chem. 2012, 54, 22-32.
- Bogolubsky, A. V.; Moroz, Y. S.; Mykhailiuk P. K.; Ostapchuk, E. N.; Rudnichenko, A. V.; Dmytriv, Y. V.; Bondar, A. N., Zaporozhets O. A.; Pipko, S. E.; Doroschuk, R. A.; Babichenko, L. N.; ACS Comb. Sci. 2015, 17, 348-354.
- Azam, F., Therapeutic potential of free radical scavengers in neurological disorders. Handbook of Free radicals: Formation, Types and Effects. New York: Nova Publishers 2010; 57-97.
- Kurek-Górecka, A.; Rzepecka-Stojko, A.; Górecki, M.; Stojko, J.; Sosada, M.; Świerczek-Zięba, G.; Molecules 2013, 19, 78-101.
- Khan, I.; Ali, S.; Hameed, S.; Rama, N. H.; Hussain, M. T.; Wadood, A.; Uddin, R.; Ul-Haq, Z.; Khan, A.; Ali, S.; Choudhary, M. I.; Eur. J. Med. Chem. 2010, 45, 5200-5207.
- Bayoumi, W. A.; Elsayed, M. A.; Baraka, H. N.; Abou‐zeid, L.; Arch. Pharm. (Weinheim). 2012, 345, 902-910.
- Zhao, P. L.; Duan, A. N.; Zou, M.; Yang, H. K.; You, W. W.; Wu, S. G.; Bioorg. & Med. Chem. Lett. 2012, 22, 4471-4474.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


