Antagonistic Mechanism of Chalcone Derivatives Against Human Estrogen Alpha of Breast Cancer Using Molecular Dynamic Simulation
Muchtaridi Muchtaridi1 , Muhammmad Jajuli1 and Muhammad Yusuf2
, Muhammmad Jajuli1 and Muhammad Yusuf2
1Departmen of Pharmaceutical Analysis and Medicinal Chemistry, Faculty of Pharmacy, Universitas Padjadjaran, Jln. Raya Bandung Sumedang KM.21, Jatinangor 45363, West Java, Indonesia.
2Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Padjadjaran, Jln. Raya Bandung-Sumedang KM. 21, Jatinangor 45363,West Java, Indonesia.
Corresponding Author E-mail: muchtaridi@unpad.ac.id
DOI : http://dx.doi.org/10.13005/ojc/340607
Article Received on : 09-10-2018
Article Accepted on : 11-11-2018
Article Published : 17 Nov 2018
2’,4’-dihydroxy-6-methoxy-3,5-dimethylchalcone (ChalcEA) that isolated from Eugenia aquea Burm f. leaves has a potential anticancer activity against human breast adenocarcinoma cell lines (MCF-7). The objective of this study was to modify ChalcEA to increase its activity as an antagonist of breast cancer with computational simulation approach. A molecular docking simulation was done against the modification structure of ChalcEA with Autodock4 to determine binding interaction between ChalcEA and hERα receptor agonists (PDB ID 1g50). Subsequently, the structure with the smallest bond energy value from the docking result was simulated using molecular dynamics to see its stability within a certain time. The results of molecular docking showed that ChalcEA modification which has a phenol group and pyrazole (MK2) had the free binding energy (ΔG) with a value of -10.2 kcal/mol and bonding hydrogen with GLU353 and ARG394, while estradiol had a value of ΔG=-10.7 kcal/mol. Based on molecular dynamics results, the determination of binding energy was gained using MMPBSA (Molecular Mechanics Poisson-Boltzmann and Surface Area) calculation methods. The MK2 has the better affinity than estradiol with a value of ΔGTotal=-45.10 kcal/mol, while estradiol was amounted to -40.86 kcal /mol. This study suggests that the MK2 might be potential as an antagonist to the hERα of breast cancer.
KEYWORDS:Breast Cancer; Chalcone; hERα; Molecular Docking; Molecular Dynamics
Download this article as:| Copy the following to cite this article: Muchtaridi M, Jajuli M, Yusuf M. Antagonistic Mechanism of Chalcone Derivatives Against Human Estrogen Alpha of Breast Cancer Using Molecular Dynamic Simulation. Orient J Chem 2018;34(6). |
| Copy the following to cite this URL: Muchtaridi M, Jajuli M, Yusuf M. Antagonistic Mechanism of Chalcone Derivatives Against Human Estrogen Alpha of Breast Cancer Using Molecular Dynamic Simulation. Orient J Chem 2018;34(6). Available from: http://www.orientjchem.org/?p=52783 |
Introduction
Cancer is a normal cell that has an uncontrolled proliferation tissue whose differentiation rate is higher than normal cells.1 Cancer cells have angiogenesis ability1 and can spread to its nearby cells or tissues.2 Clinical symptoms of cancer is manifested when it is reaching a high stage, making it difficult to treat. A total of 8.2 million people died from cancer in 2012.3 At all times, the highest rates of death are caused by cancer of liver, lung, colorectal, stomach, and breast.3 Genderwise, in Indonesia the number of female cancer patients is larger than male; approximately 2.2:0.6 per 1000 population4
Breast cancer is caused by tumors in the breast that are sensitive to estrogen.6 Growth hormone that can trigger the development of breast cancer cells is secreted by human breast’s epithelial cells, stimulated by progesterone7 and estrogen8 Breast cancer therapy can be done by inhibiting estrogen activity or working competitively with estrogen.9
Drug discovery from medicinal plants has played an important role in the treatment of cancer and, indeed, the newest clinical applications of plant secondary metabolites and their derivatives over the last half a century have been applied in combating cancer.10
Currently, anticancer agents from plants in clinical use and can be categorized into four main classes of compounds, namely: vinca (or Catharanthus) alkaloids, epipodophyllotoxins, taxanes, and camptothecins. Vinblastine and vincristine are isolated from Catharanthus roseus (L.) G. Don (Apocynaceae) (formerly Vinca rosea L.) and have been used clinically for over 40 years.11 Vinca alkaloids and several of their semi-synthetic derivatives block mitosis with metaphase arrest by binding specifically to tubulin, resulting in its depolymerization.12 Numerous derivatives of this compound have been synthesized, some of which are currently in clinical use.12 All of these natural products have led to significant biological discoveries related to their unique mechanisms of action.
In our previous study, we evaluated 42 species of Indonesian primate-consumed plants for their antiproliferative activity against MCF-7 human breast cell lines using a MTT bioassay. The results showed that some extracts of the plants had strong inhibitory activity against MCF-7 cell proliferation, and one of them was Eugenia aquea leaves extract. Subsequently, 2’,4’-dihydroxy-6- methoxy-3,5-dimethylchalcon (ChalcEA) was isolated from Eugenia aquea leaves and further investigations were conducted to provide a basis for its use in breast cancer disease management. The results showed that the compound inhibited cell proliferation in a dose-dependent manner with IC50 of 74.5 µg/mL (250 µM) and promoted apoptosis via the activation of PARP.12
Furthermore, the 2’,4’-dihydroxy-6-methoxy-3,5-dimethylchalcon was docked against Human Estrogen Alpha (hERα), and its affinity was compared to 4-hydroxytamoxifen (4-OHT). However, this compound could not compete with tamoxisfen due to its lack of hydrophobic tail. A chemical compound, namely chalcon, isolated from E. aquea leaves was then analyzed for their possible Selective Estrogen Reseptor Modulator (SERM)-like properties by using a series of appropriate cell systems/markers reflective of SERM activity. Their action was assessed in induction of human breast cancer cell lines which are estrogen dependent markers in MCF-7 breast cancer cells.
Chalcone is a natural compound that includes flavonoid precursors and isoflavonoid, which is also widely found in plants. Having two open-chain flavonoids, there are two aromatic rings combined by three α, β-unsaturated carbons.14 Chalcone has biological activities including antioxidant,15 antibacterial,16 anti-inflammatory,17 and anti-cancer.18
Chalcone has a structural similarity with estradiol and tamoxifen. This causes chalcone to have estrogenic ability (agonist) and antiestrogen (antagonist),19 Thus, this study used estrogen α receptors (hER-α) in the agonist complex with estradiol ligand. The ChalcEA has activity-based apoptotic activity on PARP (poly- (ribon) polymerase) and proliferative inhibition cells in a given dose with IC50 74.5 μg/mL (250 μM).10 The IC50 value of inhibitory activity of cell proliferation belongs to the not very active category with a range of 10-100 μg/mL.20 Therefore, this study modified ChalcEA to increase its affinity to estrogen receptor.
Materials and Methods
Materials
Software
This study used software namely: ChemBioDraw Ultra 12.0 and ChemBio3D Ultra 12.0 (PerkinElmer Inc), BIOVIA Discovery Studio Visualizer 2016 (Accelrys Inc., San Diego, CA, USA), Autodock 4.2 (The Scripps Research Institute), and AMBER 14 (Amber, San Francisco, CA, USA).
Methods
Preparation of Protein Receptor
The hER-α receptor complex was obtained from Protein Data Bank with ID pdb 1g50, which had been complexed with estradiol. Using BIOVIA Discovery Studio Visualizer 2016. The estradiol structure was separated as a standard ligand and water molecules are removed to obtain only the hERα receptor.
Preparation of Ligands
Ligands were obtained by drawing the 2D structures from chalcone derivatives using Chem Bio Draw Ultra 12.0 and stored using the “.mol format”. This format was then run on ChemBio3D Ultra 12.0 to minimize and add charge. Afterward, it was stored in pdb format in order to obtain its 3D ligand structure. The estradiol structure was then used for validation process.
Molecular Docking Simulation
This method was modified from our previous study.19 The repetitions with 100 run of molecular docking were performed using GA parameters as scoring function.19 The search area was limited by grid box dimensions of x=104.963, y=14.909, and z=23.535 with a volume of 40x40x40 points. Docking parameters were afterwards saved in “.dpf format”. Subsequently, the automatic docking process was run. The results of the docking process were stored in “.dlg format”. In this data, there were bond energy values (EI), inhibition constants (KI), and ligand conformations. The best ligand conformation with the smallest binding energy value could be visualized using BIOVIA Discovery Studio Visualizer 2016.
Molecular Dynamics (MD) Simulation
The simulation was performed on two systems, namely hERα 1g50-estradiol (agonist) and 1g50-derived ChalcEA which have the best affinity. At AMBER 14, the ligand was parameterized using Antechamber with AM1-BCC calculations. Then, Cl– ion was added as counter ion using LEAP. The disolvation system with added water as solvent was explicitly a water model TIP3P box.
The minimization of each complex was performed with the aid of Sander module in AMBER 14. The minimization can remove clusters or atoms that will cause collisions between atoms or eliminate steric hindrances. This contact was removed to maintain low energy within the area during the simulation. Then, the minimization was conducted on the heating complex gradually from 0-100K, 100-200K, and 200-310K with harmonic restraint of 5 kcal.mol-1· A-2 on the backbone atoms. Furthermore, MD simulation was done for 20 ns with constant temperature of 310K.
Results
Molecular Docking Simulation
The results of docking estradiol and modification of ChalcEA can be seen in Table 1. These results showed that MK1 and MK2 have better affinity for hER-α (1g50) than ChalcEA. The structures of MK1 and MK2 can be seen in Figure 1, and their interaction with hER-α can be seen in Figure 2. The other molecular structure of ChalcEA modifications (MK) can be seen in the supplementary file.
Table 1: Results of Estradiol Docking and Chalcone Modification of hER-α (PDB ID 1g50).
| No. | Compound code | ΔG1 (kcal/mol) | Molecular weight | Ki2 (uM) |
| 1 | Estradiol | -10.7 | 272.38 | 13.23×10-3 |
| 2 | CHalcEA | -8.2 | 298.33 | 897.61×10-3 |
| 3 | MK1 | -10 | 476.56 | 42.41×10-3 |
| 4 | MK2 | -10.2 | 434.48 | 29.02×10-3 |
| 5 | MK3 | -9.2 | 466.57 | 160.63×10-3 |
| 6 | MK4 | -8.3 | 449.58 | 822.33×10-3 |
| 7 | MK5 | -8 | 471.54 | 1.33 |
| 8 | MK6 | -7.5 | 447.52 | 2.73 |
| 9 | MK7 | -7.5 | 473.52 | 3.02 |
| 10 | MK8 | -5.9 | 440.49 | 43.47 |
| 11 | MK9 | -5.6 | 463.52 | 74.21 |
| 12 | MK10 | -4.2 | 473.52 | 799.7 |
| 13 | MK11 | -2.6 | 501.62 | 12.19×103 |
1ΔG: Free energy binding of molecular docking simulation results
2Ki: Inhibition constant that calculated based on the equation: Ki = exp (DG/ (R.T)
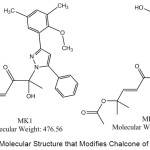 |
Figure 1: The Molecular Structure that Modifies Chalcone of MK1 and MK2. |
 |
Figure 2: MK1 (Grey) and MK2 (Green) Interactions with Human Estrogen Receptor Alpha (HER-α) Receptors Hydrogen Bond (Green Line). Click here to View figure |
Molecular Dynamics (MD) Simulation
Based on Figure 3, the system generally achieves stability after reaching frame 2000 (2 ns). The MK2-receptor complex has a high RMSD compared to the receptor complex with estradiol. Visible high deviation on Res-MK2 achieved at 4000 frames (4 ns) thereafter is relatively stable with a value of ~2.5 Ǻ.
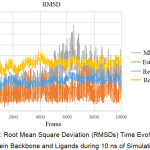 |
Figure 3: Root Mean Square Deviation (RMSDs) Time Evolution of Protein Backbone and Ligands during 10 ns of Simulation. |
RMSF (Root Mean Square Fluctuation)
The RMSF value in Figure 4 below shows fluctuations for each residue during simulation, displaying the flexibility of the residue. The RMSF value of each residue can be seen in Figure 4 below.
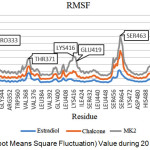 |
Figure 4: RMSF (Root Means Square Fluctuation) Value during 20 ns Simulation Time. |
The MK2 ligand disrupts the stability of the hydrogen bond with increasing fluctuations in GLU419. As seen in Figure 5, in the complex with MK2, loop-534 is elevated compared to estradiol.
 |
Figure 5: Comparison loop-534 of MK2 (Orange) Complex is More Elevated than Estradiol (White). Loop-534 is the region between Helix-11 and Helix-12 that accomadate antagonist compounds.21,23 |
According to Table 2, the overall MK2 system has a lower energy value than estradiol. In both systems, van der walls (vdw) energy has the smallest value compared to other energies in both systems.
Table 2: Energy Calculation Binding of MMPBSA Method.
| Energy (kcal/mol) | System1 | |
| MK2- hERα | Estradiol- hERα | |
| Vdw2 | -57.06 | -44.87 |
| EEL3 | -9.63 | 1.16 |
| EGB3 | 29.36 | 7.39 |
| ESURF3 | -7.78 | -4.54 |
| ΔGgas (vdw + EEL) | -66.69 | -43.71 |
| ΔGsolv (EGB + ESURF) | 21.58 | 2.85 |
| ΔGTotal | -45.1 | -40.86 |
1System of molecular dynamic that run with time space 20 ns
2Vdw: Van der Waals interaction energy,
3EEL: Electrostatic energy, EGB:, ESURF: Surface area energy, ΔGsolv: solvation free energy.
Discussion
X-ray structure of ERα that had been complexed with 4-OHT (PDB ID: 3ERT) was taken based on a good experimental resolution (1.9 Å), R-value free (0.262), and R-value work (0.229).22 4-OHT has hydrophobic interaction that predominantly interacts with aromatic rings. The butenyl group also forms a positive ionisable interaction with secondary amine nitrogen of hERα. The hydroxyl and phenoxy oxygen formed hydrogen bond interactions with GLU353. Helices 3, 6, 7, 8, 11, and 12 were observed, as they played an important role. Thus, ERα has ligand-binding domain (LBD) which was predominantly the hydrophobic cavity. The interaction of ligand with 536-544 residues of Helix-12 in macromolecule (ERα) has a role to determine whether the ligand is agonist or antagonist. An antagonist ligand binds to LBD of ERα. Thus, the Helix-12 will be closed and does not bind to co-activator. This interaction causes lack of hydrogen bond network called ‘zipper’ network in agonist structure. This network was built between GLU419, HIS524, and LYS531 that has an important role in ensuring that Helix-3 and Helix-11 are in close contact.21
Based on the previous study,23 the modification was based on the hydrophobicity of the functional group because hER-α receptor has a hydrophobic environment and refers to the estradiol, including steroids. Tamoxifen is a prodrug widely used as analog antagonist estrogen. The 4-hydroxytamoxifen (4-OHT) compound is a tamoxifen metabolite. The ChalcEA had no hydrophobic tail as in 4-OHT, but only carbonyl group. In addition, the ChalcEA carbonyl group was shorter than the diethylaminoethoxy group at 4-OHT and was responsible for its affinity. ChalcEA derivatives with pyrazole rings have a significant activity in inhibiting human cancer cell line proliferation.19 It can be used as a basis in modifying ChalcEA in this study (Table 1).
Estrogen receptors have a hydrophobic environment that composes Helices 3, 6, 7, 8, 11, and 12. Helix-12 has an important role in determining ligand activity as an agonist or antagonist. Helix-12 can bind to the active side of the co-activator to be an agonist. Helix-12 composers include the His524 residue.21 MK1 and MK2, on the other hand, do not form this bond, so it is antagonistic. Then, hydrophobic tail is added to the structure of MK1 and MK2, thus increasing the hydrophobicity of the ligand. This tail is based on an anti-breast cancer from commercial synthetic known as tamoxifen as has been referred to on our previous study.
MK2 forms hydrogen bonding interactions with GLU353 and ARG394 residues with a distance of 2 Ǻ. The distance is included in strong hydrogen bonds. In addition, it causes the formation of T-shaped pi-pi interaction. This interaction is formed between the phenol rings with PHE404, while MK1 only forms hydrophobic interaction. With the formation of hydrogen bonds, MK2 is more stable in hER-α complex compared with MK1. Then, to observe the behaviour of complex MK2 further, MD simulation was conducted. MK2 was chosen because it has a smaller bond energy value and it can bind to the same residue as estradiol at hERα receptors (GLU353 and ARG394). The MD simulation was performed on two systems, namely hERα 1g50-estradiol (agonist) and 1g50-MK2.
The first stage in the MD simulation was that hER- α receptor was first given a force field with LEAP program. Protein was added to force field AMBER FF14SB. Force fields can improve accuracy for protein chain parameters.24 Then, the ligand was parameterized to calculate the charge from the constituent atom using semi-empirical quantum AM1-BCC calculation minimization method.
The MD simulation was done in ten stages. Each stage took place in a simulation time of 2 ns, making the overall time simulation of 20 ns. The data generated from the MD simulation included RMSD (Root Mean Square Deviation) and RMSF (Root Mean Square Fluctuation). These data illustrated the complex behaviour of ligand-receptors in the presence of pressure and temperature. Each of these data was obtained after a simulation of 20 ns. The RMSDs time evolution of the protein backbone and ligands during 10 ns of simulation are shown in Figure 3. This suggests that the receptor flexibility in MK2 ligands increases, compared to estradiol complexes, with lower flexibility and a sharply increased RMSD in frame 2000 with ~1.5 Ǻ. This increase in RMSD values marks the opening of the receptor structure (unfold).
Based on Figure 4, it is generally possible to see the receptor’s flexibility in the same residue. The influence of the ligand also affects the flexibility of the receptor residue. MK2 ligands show higher fluctuations compared with ChalcEA and estradiol, while on estradiol complex the resulting fluctuation is lower. Residues with high fluctuations in each ligand are PRO333, THR371, and SER463, whereas in GLU419 only on MK2 ligand shows high fluctuation. This fluctuation increase was caused by a disruption of hydrogen bond by GLU419. In the crystal structure, PDB 1g50 is known to have a series of hydrogen bonds formed between GLU419 with LYS531 and GLU419 with HIS524. The disturbance of hydrogen bonding to the agonist receptor structure (1g50) may direct the ligand to have antagonistic action.21
This interaction caused lack of hydrogen bond network called ‘zipper’ network in agonist structure. This network was built between GLU419, HIS524, and LYS531 that has play an important role in ensuring that Helix-3 and Helix-11 are in close contact [23]. In Helix-3 and Helix-11, loop-534 in the complex with MK2 is more open than the estradiol agonist complex due to the opening of hydrogen bonding interaction. Then MK2 is suspected to having an action as an ER antagonist.
Vdw energy of MK2 is smaller, due to the structure of the larger MK2. This shows that in the constituents of the active pockets of hERα, there are many residues that are hydrophobic. MK2 thus has a better affinity for hER-α than estradiol, with ΔGTotal smaller than estradiol.
Conclusion
MK2 has the potential as an antagonist of receptor hERα breast cancer. Based on the results of the docking simulation, it has a value of ΔG=-10.2 kcal/mol, occupying the hERα binding site by forming hydrogen bond GLU353 and ARG394. Based on the MD results, it can be seen that MK2 has a better affinity than estradiol with ΔGTotal=-45.10 kcal/mol, while estradiol is -40.86 kcal/mol.
Conflicts of Interest
There is no conflict of interest.
Acknowledgements
This study was supported by The Directorate General of Higher Education of The Ministry of Research and Technology of the Republic of Indonesia through the International Research Collaborations and Publications Grants No. 391k/UN6.0/LT/2018.
References
- De Berardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C. B. Cell Metab. 2008, 7,11-20.
CrossRef - Goustin, A. S.; Leof, E. B.; Shipley, G. D.; Moses, H. L. Cancer Res. 1986, 46,1015-29.
- Ministry of Health of Indonesia. Data and Information on Indonesia Health Profile. Jakarta: Ministry of Health Republic of Indonesia; 2015:2-3.
- RI KK. Riset Kesehatan Dasar (RISKESDAS). Jakarta: Badan Litbang Kemenkes RI, 2013.
- Lombardi, S.; Honeth, G.; Ginestier, C.; Shinomiya, I.; Marlow, R.; Buchupalli, B.; Gazinska, P.; Brown, J.; Catchpole, S.; Liu, S.; Barkan, A.; Wicha, M.;Purushotham, A.; Burchell, J.; Pinder, S.; Dontu, G. Stem Cell Reports , 2014,2,780-793.
CrossRef - D. J. Jerry, J. D. Shull, D. L. Hadsell, M. Rijnkels, K. A. Dunphy, S. S. Schneider, et al.,. Mamm Genome, 2018. 29, 24-37.
CrossRef - Russo, J.; Russo, I. H. J Steroid Biochem Mol Biol. 2006, 102,89-96.
CrossRef - Butler, M. S.. J Nat Prod. 2004,67, 2141-2153.
CrossRef - Lumachi, F.; Santeufemia, D. A.; Basso, S. M. M. World Journal of Biological Chemistry 2015, 6 (3), 231-239.
CrossRef - Van Der Heijden, R.; Jacobs, D. I.; Snoeijer, W.; Hallard, D.; Verpoorte, R. Curr. Med. Chem, 2004, 11, 607-628.
CrossRef - Okouneva, T.; Hill, B. T.; Wilson, L.; Jordan, M. A. Mol Canc Thers. 2003, 2, 427-436.
- Subarnas, A.; Diantini, A.; Abdulah, R.; Zuhrotun, A.; Hadisaputri, Y. E.; Puspitasari, I.M.; Yamazaki, C.; Kuwano, H.; Koyama, H. Oncol Lett. 2015, 9,2303-2306.
CrossRef - Syam, S.; Abdelwahab, S.I.; Al-Mamary, M.A.; Mohan, S. Molecules. 2012, 17,6179.
CrossRef - Vogel, S.; Ohmayer, S.; Brunner, G.; Heilmann, J. Bioorg Med Chem. 2008, 16, 4286-93.
CrossRef - Božić, D.D.; Milenković, M.; Ivković, B.; Ćirković, I. Indian J Med Res. 2014, 140, 130-137.
- Cheng, J.H.; Hung, C.F.; Yang, S.C.; Wang, J.P.; Won, S.J.; Lin, C.N. Bioorg Med Chem. 2008,16, 7270-7276.
CrossRef - Tamir, S.; Eizenberg, M.; Somjen, D.; Izrael, S.; Vaya, J. The J. Steroid Biochem Mol Bio. 2001, 78, 291-98.
CrossRef - Weerapreeyakul, N.; Nonpunya, A.; Barusrux, S.; Thitimetharoch, T.; Sripanidkulchai, B. Chin Med. 2012, 7, 15.
CrossRef - Muchtaridi, M.; Syahidah, H.N.; Subarnas, A.; Yusuf, M.; Bryant, S. D.; Langer, T. Pharmaceuticals. 2017, 10, 1-12.
CrossRef - Jones, G.; Willett, P.; Glen, R. C.; Leach, A.R.; Taylor, R. J Mol Biol. 1997, 267,727-48.
CrossRef - Shiau, A. K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. Cell. 1998, 95,927–37.
CrossRef - Hawash, M.M.; Kahraman, D.C.; Eren, F.; Cetin Atalay, R.; Baytas, S.N. Eur J Med Chem. 2017,129, 12-26.
CrossRef - Muchtaridi. M.; Yusuf, M.; Diantini, A.; Choi, S.B.; Al-Najjar, B.O.; Manurung, J.V.; Subarnas, A.; Achmad, T.H.; Wardhani, S.R.; Wahab, H.A. Int J Mol Sci. 2014,15,7225–49.
CrossRef - Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, KE.; Simmerling C. J Chem Theory Comput. 2015,11,3696-713.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


