Studies on Photophysical Properties of Mono-Carbonyl Curcumin Analogues: Experimental and Theoretical Approach
Manjula Rayanal, Prasad Pralhad Pujar and Sridhar D
and Sridhar D
Department of Chemistry, Christ (Deemed to be University), Bengaluru – 560 029, India.
Corresponding Author E-mail: prasad.pujar@christuniversity.in
DOI : http://dx.doi.org/10.13005/ojc/3404059
Article Received on : 17-03-2018
Article Accepted on : 30-05-2018
Article Published : 19 Jul 2018
The solvatochromic fluorescence behaviour of mono-carbonyl curcumin analogues has been studied in ten different solvents ranging from non-polar to polar. The solvent effect on the spectral properties of analogues has been discussed. The ground state dipole moments were estimated experimentally by Bilot-Kawski equation which is a function of Stokes shift with the solvent polarity parameters and Guggenheim method and theoretically by TD-DFT studies. The excited state dipole moment was determined using Bilot-Kawski equations. The excited state dipole moments for the two molecules were found to be higher than their corresponding ground state dipole moments. Theoretically Frontier molecular orbital (HOMO/ LUMO) energies were determined by Gaussian 09 W software using TD-DFT.
KEYWORDS:Curcumin; Dipole Moment; HOMO/LUMO Energies; TD-DFT
Download this article as:| Copy the following to cite this article: Rayanal M, Pujar P. P, Sridhar D. Studies on Photophysical Properties of Mono-Carbonyl Curcumin Analogues: Experimental and Theoretical Approach. Orient J Chem 2018;34(4). |
| Copy the following to cite this URL: Rayanal M, Pujar P. P, Sridhar D. Studies on Photophysical Properties of Mono-Carbonyl Curcumin Analogues: Experimental and Theoretical Approach. Orient J Chem 2018;34(4). Available from: http://www.orientjchem.org/?p=47387 |
Introduction
The golden spice curcumin is a hydrophobic natural product present in turmeric, a yellow active component isolated from plant curcuma longa L,1-3 which has been used for ages as a spice, cosmetics and in traditional medicines.4 Curcumin, its derivatives and analogues have shown extensive range of beneficial activities including antimicrobial, anticancer,5-9 antioxidant,10 anti-inflammatory,11,12 anti-tumour,13,14 anti-malarial,15 anti-mutagenic, free radical scavenging activity,16 anti-carcinogenic activity, cytotoxic activity17 and also found to show cyclooxygenase18 and tubulin polymerization inhibitory activities.19
In addition to the biological and pharmacological application of curcumin, its electronic absorption and fluorescence properties are of extraordinary significance for analytical and molecular scale functional and structural studies. The variation of absorption, fluorescence properties with nature of solvent has given important information on the interaction of curcumin with proteins such as liposomes and human albumin20 etc. Curcumin and its analogues exhibit a marked solvent dependence on the electronic absorption and emission spectra. The photo physical properties of curcumin analogues are widely explored using absorption and fluorescence study in various solvents.20-29
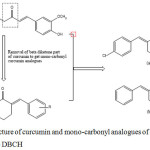 |
Figure 1: Structure of curcumin and mono-carbonyl analogues of curcumin (a) CBCH and (b) DBCH |
The Solvatochromism describes the strong reliance of absorption and fluorescence spectra on different solvents. The spectral behaviour of organic compound is directly related to the electronic structure of the solute and solvent molecules. A change in the nature of solvent is accompanied by a change in polarity, dielectric constant and polarizability of the surrounding medium. A change in solvent polarity will cause difference in stabilization of the ground and excited states of solute molecule and hence, a change in the energy gap between ground state and the excited state, accordingly the difference in the shape, position and intensity of the emission and absorption spectra.
The information of ground state and excited state dipole moments is useful in understanding the electronic and geometric structures of organic compounds and in designing new molecules with non-linear optical characters and in determining the progress of a photochemical conversion.30 The excited state dipole moment of organic compounds is also useful in determining the tunability range of their emission energies using polarities of the solvent.
In the present study, we have demonstrated the effects of different solvents on electronic absorption and fluorescence spectra of two biologically active curcumin analogues viz, 2,6 bis(4-chlorobenzylidene) cyclohexanone (CBCH) and 2,6-dibenzylidenecyclohexanone (DBCH). Further, the ground state dipole moment is experimentally calculated using Bilot-Kawski method and Guggenheim method and theoretically by TD-DFT studies and excited state dipole moment was estimated using Bilot-Kawski approach. Computational analysis of mono-carbonyl curcumin analogues were done to supplement the experimentally determined values.
Experimental
Curcumin analogues viz, CBCH and DBCH were prepared as given in the literature.31 The structure and purity of analogues were confirmed by NMR spectra and melting point. The structures of CBCH and DBCH are presented in Fig 1. In the current investigations we have used ten solvents which are hexane, toluene, diethyl ether, chloroform, dichloromethane, acetone, ethyl acetate, acetonitrile, ethanol and methanol.
Absorption spectra were recorded using Shimadzu UV-visible spectrophotometer (UV-visible 1800). The fluorescence spectra were recorded using Shimadzu spectrofluorometer (RF5301FC). All these measurements were recorded in very dilute solutions at room temperature. Origin 8.0 Professional program was used to analyse solvatochromic results. All the chemicals used were purchased from SD Fine Chemicals Ltd.
Experimental Calculation of Ground and Excited State Dipole Moments
The excited state dipole moment of the organic compound is calculated using the effect of internal or external electric field on its spectral band positions. To calculate the excited state dipole moments spectral shifts of absorption and emission in different solvents are used. Dipole moments of the mono-carbonyl analogues have been calculated using two methods. Method I contains calculation of the ground state and excited state dipole moments using Bilot-Kawski approach and Method II contains the estimation of the ground state dipole moments using Guggenheim method.
Method I
In the first method the ground state and excited state dipole moments was calculated using Bilot-Kawskiapproach32,33 which is a function of Stokes shift with parameters which dependence on refractive index and dielectric constant of the solvent molecule.
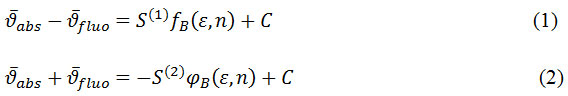
Where
![]()
Where
![]()
absorption maxima and
![]()
fluorescence maxima in wavenumber (cm-1), ε represents the dielectric constant, n represents the refractive index of different solvents and C is integration constant. fB (ε,n) and gB (n) are solvent polarity parameters based on refractive index and dielectric constant.
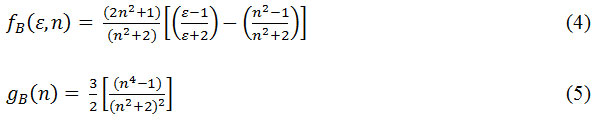
The plots of
![]()
against fB (ε,n)
and
![]()
Against φB (ε,n) gives a linear plot with respective slopes S(1) and S(2). These slopes are given by

Where µgs – the ground state and µex– the excited state dipole moment, h-Planck’s constant, a-Onsagers cavity radius and c-velocity of light. The value of the Onsager’s cavity radius for both compounds were estimated using Suppan’s equation34
![]()
Where M, N, d represents the molecular weight, Avogadro’s number and d density (assumed to be 1 g cm-3)
The change of the dipole moment during transition is expressed as

Solvatochromic shift can be important in determining the type of interaction between the solute molecule and nature of solvent and in estimating the dipole moments of organic compounds in the ground state and in the excited state. Assuming the ground state and excited state dipole moments are parallel.35
The ground state and excited state dipole moments were estimated using following equations.
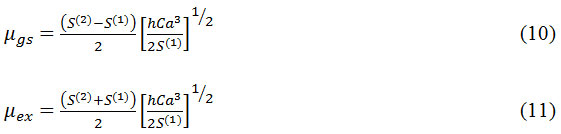
The ratio of excited dipole moment to the ground state dipole moment is given by
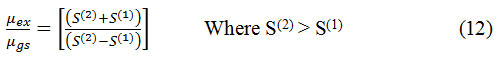
When µgs and µex are not parallel than the angle φ is formed between them and that can be estimated by following equation (13)
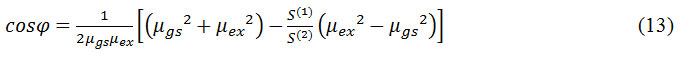
Method II
The ground state dipole moment (µgs) is calculated from the dielectric measurements in dilute solutions using number of methods. One of such method is Guggenheim method.36
In Guggenheim method µg is estimated using following expression (14)

Where Δ = (D12 – η12 2 ) -(D1 – η12)
k-Boltzmann constant, T-Absolute temperature and N- Avogadro’s number, C- concentration of the compound in given solvent and D1, D12 represents the dielectric constant of solvent and solution and η1 and η12 represents the refractive index of the solvent and that of solution.
The dielectric constant of the dilute solutions are estimated by determining the capacitance of the solution using the equation (15)
![]()
Where Cs – capacitance due to leads, Cx – capacitance of solution and Ca – capacitance in air.
Results and Discussion
Solvent Effect on the Absorption Spectra
Absorption spectra of the mono-carbonyl curcumin analogues were recorded in ten different organic solvents varying from non-polar to polar at room temperature. In our study, the absorption spectrum of CBCH and DBCH were recorded in solvents with dielectric constants varying from 1.88 to 37.5. The absorption maximum of CBCH in ten different solvents was obtained in the range of 325 nm to 331 nm and for DBCH in the range 324 nm to 330 nm (Fig 2). It has been observed that shift in absorption maximum was very small. The smaller shift in the electronic absorption spectrum with varying solvent polarity, suggests that the energy distribution in the ground state is not affected to a larger extent. This is may be due to the ground state of CBCH and DBCH are less polar than the excited state.
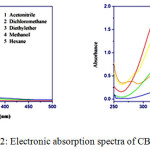 |
Figure 2: Electronic absorption spectra of CBCH and DBCH |
Solvent Effect on the Emission Spectra
The fluorescence spectra of CBCH and DBCH were recorded in ten different organic solvents of varying solvent polarities. Stokes shifts were calculated and tabulated in Table 2 and solvent polarity parameters are depicted in Table 1. For the curcumin analogues, CBCH and DBCH when we increase the solvent polarity from non-polar to polar, the emission spectra was shifted to the longer wavelength from 430 nm to 603 nm i.e. fluorescence spectra exhibits large band sensitivity to the medium effect. The emission spectra of CBCH and DBCH are displayed in Fig 3. The bathochromic shift from 328 nm to 603 nm corresponding to 275 nm shift confirms π-π* transitions which arises from conjugation of α-β unsaturated carbonyl group to aromatic ring system.
The emission spectrum shows a large shift than the absorption shift. As we increase the solvent polarity, bathochromic shift has been observed in fluorescence spectra due to the large variance in the charge distribution of molecule in the excited than in the ground state, which causes a stronger interaction of polar solvents in the excited state than in the ground state.
The value of Stokes shift varies from 6737 to 13262 cm-1 for CBCH and 7231 to 13904 cm-1 for DBCH. This indicates inter molecular charge transfer transitions. The large value of Stokes shift implies that the geometry of the molecule in the excited state may be different from that of the ground state. It has been observed that Stokes shift values increased with changing solvent polarities form non polar to polar. This suggests the charge transfer transition in CBCH and DBCH molecule resulting in increased excited state dipole moment values.
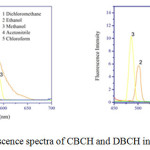 |
Figure 3: Fluorescence spectra of CBCH and DBCH in different solvents. |
Table 1: Solvent polarity functions for Bilot-Kawski method.
| Solvents | fB (ε,η) | gB (η) | φB (ε,η) |
| n-Hexane | 0.0240 | 0.2550 | 0.5126 |
| Toluene | 0.0310 | 0.3289 | 0.6917 |
| Diethyl ether | 0.3790 | 0.2457 | 0.8704 |
| Chloroform | 0.3720 | 0.3022 | 0.9753 |
| Ethyl acetate | 0.4930 | 0.2530 | 0.9958 |
| Dichloromethane | 0.5952 | 0.2877 | 1.1703 |
| Acetone | 0.7927 | 0.2443 | 1.2814 |
| Ethanol | 0.8169 | 0.2457 | 1.3083 |
| Methanol | 0.8870 | 0.2241 | 1.3041 |
| Acetonitrile | 0.8630 | 0.2342 | 1.3310 |
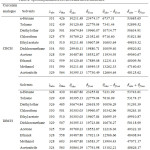 |
Table 2: Spectral parameters of CBCH and DBCH molecules. |
Estimation of Ground State and Excited State Dipole Moments
The shifts of emission maxima on changing the solvent polarities are more prominent than the shifts of absorption maxima. This implies that change in dipole moments (Δµ) is positive and dipole moment of both compounds increase on excitation. According to Bilot-Kawski approach, the plots of
![]()
With f B (ε,η) and
![]()
with φB (ε,η) for CBCH and DBCH molecules have given linear plots with good correlation coefficient.
For CBCH molecule the plot of
![]()
with f B (ε,η) has given a good correlation coefficient (0.9245) with slope S(1) 6457.19 and intercept at 7134.01 and the graph
![]()
with φB (ε,η) has also given good correlation coefficient 0.8788 and slope S(2) 7147.67 and intercept at 57564.66. The values of slope S(1) and S(2) are used to calculate the ground and excited state dipole moments of CBCH using equations (10) and (11) and these were found to be µgs=0.5268 and µex=10.379. All these values are displayed in (Fig 4 Table 3, 4). Using µgs and µex the change in dipole moment and the ratio of µgs and µex has been calculated.
For DBCH molecule the plot
![]()
with f B (ε,η) has given graph with most of the points were fitted linearly with good correlation of 0.96307 and corresponding slope S(1) 6678.02 and intercept at 7331.84 and the plot
![]()
with φB (ε,η) the correlation coefficient was 0.8837 with slope S(2) 6887.92 and intercept at 57384.86 was obtained (Fig 5). Using the values of slopes S(1) and S(2) the dipole moments of DBCH was calculated by using equation (10) and (11) and these values are equal to µgs= 0.1407 and µex=9.099. All these values are presented in Table 3 and 4. Using µgs and µex the change in dipole moment and the ratio of µgs and µex has been calculated.
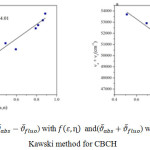 |
Figure 4 |
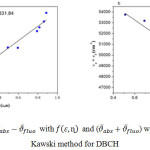 |
Figure 5 |
Table 3: Statastical prameters obtained for Bilot-Kawski approach
| Curcumin analogues | Slope | R2 | Number of data |
| CBCH(1)DBCH(1) | 6457.196678.02 | 0.92450.9630 | 1010 |
| CBCH(2)DBCH(2) | -7147.67-6887.92 | 0.87880.8837 | 1010 |
Statastical parameters obtained for the graph of
![]()
with f B (ε,η) Statistical parameters obtained for the graph of
![]()
with φ (ε,η)
In method II
We have calculated the ground state dipole moments of CBCH and DBCH using Guggenheim method using equation 14 and all the values of dipole moments are summarized in Table 4.
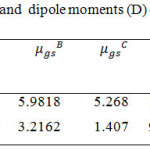 |
Table 4: Onsagers radius(A0) and dipole moments (D) of CBCH and DBCH |
Debye(D) = 3.33564 x 10-30Cm = 10-18 esu cm.
µgs = Ground state dipole moments
µex = Excited state dipole moments
AGaussian 09 W Software.
BGuggenheim method.
CBilot-Kawski equation.
DBilot-Kawski equation.
EThe µe / µg is calculated from equation (12).
FThe Δµ calculated from Bilot-Kawski method.
For CBCH molecule the ground state dipole moment calculated using Guggenheim method was equal to µgs = 5.9818 D, Gaussian 09W software was equal to µgs = 5.261 D and Bilot-kawski equation was equal to µgs = 5.268 D and the excited state dipole moment calculated by Bilot-Kawski method was equal to µex = 10.379 D. The excited state dipole moment of CBCH was found to be greater than its ground state dipole moment. This indicates the more polar nature of CBCH in its excited state and also indicates the better solute-solvent interaction in the excited state.
For DBCH molecule the ground state dipole moment calculated using Guggenheim method was equal to µgs = 3.2162 D, Gaussian 09W software was equal to µgs = 2.784 D and Bilot-kawski equation was equal to µgs = 1.407 D and the excited state dipole moment calculated by Bilot-Kawski method was equal to µex = 9.099 D. The excited state dipole moment of DBCH was found to be greater than its ground state dipole moment. This indicates the more polar nature of DBCH in its excited state and also indicates the better solute-solvent interaction in the excited state.
When the molecules CBCH and DBCH are excited the charge transfer occurs from the H atom of the phenyl ring to O atom of the ketone group. Both electron donar and acceptor are present in the same molecule. When the molecule absorbs the energy interms of photon the migration of electron from the H atom of phenyl group to the O atom of the ketone group takes place. This indicates the intramolecular charge transfer transition and implies that both CBCH and DBCH are more polar in their excited state.
Computational Analysis
The energies of HOMO and LUMO and ground state dipole moments of CBCH and DBCH were determined using time dependent density functional theory (TD-DFT). The computations were done using Gaussian 09W software. The 3D structures of frontier molecular orbitals are displayed in Fig 6 and 7. The intermolecular charge transfer can be estimated using the frontier molecular orbital energies. The energies of HOMO and LUMO of CBCH and DBCH were estimated which explains the direction of charge transfer in the molecule. The HOMO & LUMO of CBCH were equal to -5.0689 eV and -1.1202 eV and that of DBCH were equal to -4.714 eV and -2.062 eV respectively. The band gap between HOMO-LUMO is calculated and it was found to be 3.9487 eV for CBCH and 2.654 eV for DBCH. The small energy values of HOMO-LUMO energy gap indicate easier π-π* transition of electron from HOMO to LUMO. Therefore both CBCH and DBCH molecules are highly reactive.
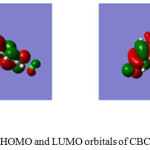 |
Figure 6: HOMO and LUMO orbitals of CBCH molecule |
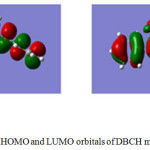 |
Figure 7: HOMO and LUMO orbitals of DBCH molecule |
The study of frontier molecular orbitals gives important information on softness and hardness of organic compounds. If the energy gap between HOMO and LUMO is less then such molecules are soft and more reactive whereas if the energy gap between HOMO-LUMO is more then such molecules are hard and less reactive.37 The chemical hardness (η) can be calculated using following equation (16).
![]()
Where EH – energy of HOMO and EL – energy of LUMO. The hardness of the CBCH is equal to η = 2.357 eV and DBCH is equal to η = 1.031 eV. We obtained chemical softness (S) using the value of chemical hardness from equation (17)
![]()
The chemical softness values for these molecules were equal to S = 0.4242 for CBCH and S = 0.9699 for DBCH. The small values of chemical hardness imply that these molecules are soft and more reactive.
The chemical potential (µ) can be calculated using the frontier molecular orbital energies using following empirical formula
![]()
The chemical potential value for CBCH molecule is found to be equal to 3.0945 and for DBCH molecule µ = 3.388.
Conclusion
In summary, the solvent effects on absorption and emission spectra and the estimated dipole moments of biologically active mono-carbonyl curcumin analogues CBCH and DBCH have been discussed in detail. A red shift was observed on increasing the polarities of the solvent indicative of π-π* transition. The ground state dipole moment has been determined theoretically using Gaussian 09 W software and experimentally by Bilot-Kawski method and Guggenheim method and the experimental excited state dipole moment was calculated using Bilot-Kawski method. It has been observed that the excited state dipole moment values are higher than the ground state dipole moment values for both the molecules CBCH and DBCH. This implies that both the molecules CBCH and DBCH are polar in the excited state than in the ground state. We also determined HOMO/LUMO energies theoretically by TD-DFT study. Based on these observations, CBCH and DBCH can be considered as good candidates for designing non-linear optical materials and fluorescent probes.
Acknowledgement
We wish to thank Anitha Varghese and K.B Akshaya, Department of chemistry, Christ university for their valuable advice and support. We thank department of IPC, IISc Bengaluru for computational analysis and would like to thank CUSAT-SAIF, Cochin University, Kochi for 1HNMR characterization.
References
- Hatcher, H.; Planalp, R.; Cho, J,; Torti, F.M.; Torti, S.V. Cell. Mol. Life Sci. 2008, 65, 1631–1652.
CrossRef - Zhan, P. Y.; Zeng, X. H.; Zhang, H. M.; Li, H. H. Food Chem. 2011, 129, 700–703.
CrossRef - Paulucci, V. P.; Couto, R. O.; Teixeira, C. C. C.; Freitas, L. A. P. Brazilian J. Pharmacogn. 2013, 23, 94–100.
CrossRef - Mague, J. T.; Alworth, W. L.; Payton, F. L. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2004, 60, 608–610.
CrossRef - Samaan, N.; Zhong, Q.; Fernandez, Z.; Chen, G.; Hussain, A. M.; Zheng,S.; Wang,G.; Chen, Q. H. Eur. J. Med. Chem. 2014, 75, 123–131.
CrossRef - Qiu, C.; Hu, Y.; Wu, K.; Yang, K.; Wang, N.; Ma, Y.; Zhu, H.; Zhang, Y.; Zhou, Y.; Chen, C.; Li, S.; Fu, L.; Zhang, X.; Liu, Z. Bioorganic Med. Chem. Lett. 2016, 26, 5971–5976.
CrossRef - Sufi, S. A.; Adigopula, L. N.; Syed, S. B.; Mukherjee, V.; Coumar, M. S.; Rao, H. S. P.; Rajagopalan, R. Biomed. Pharmacother. 2017, 85, 389–398.
CrossRef - Ohori, H.; Yamakoshi, H.; Tomizawa, M.; Shibuya, M.; Kakudo, Y.; Takahashi, A.; Takahashi, S.; Kato, S.; Suzuki, T.; Ishioka, C.; wabuchi, Y.; Shibata, H. Mol. Cancer Ther. 2006, 5, 2563–2571.
CrossRef - Fawzy, I. M.; Youssef, K. M.; Ismail, N. S. M.; Gullbo, J.; Abouzid, K. A. M. Futur. J. Pharm. Sci. 2015, 1, 22–31.
- Selvam, C.; Jachak, S. M.; Thilagavathi, R.; Chakraborti, A. K. Bioorganic Med. Chem. Lett. 2005, 15, 1793–1797.
CrossRef - Liang, G.; Shao, L.; Wang, Y.; Zhao, C.; Chu, Y.; Xiao, J.; Zhao, Y.; Li, X.; Yang, S. Bioorganic Med. Chem. 2009, 17, 2623–2631.
CrossRef - Liang, G.; Yang, S.; Zhou, H.; Shao, L.; Huang, K.; Xiao, J.; Huang, Z.; Li, X. Eur. J. Med. Chem. 2009, 44, 915–919.
CrossRef - Lin, L.; Shi, Q.; Nyarko, A. K.; Bastow, K. F.; Wu, C. C.; Su C. Y.; Shih, C. C. Y.; Lee, K. H. J. Med. Chem. 2006, 49, 3963–3972..
CrossRef - Ishida, J.; Ohtsu, H.; Tachibana, Y.; Nakanishi, Y.; Bastow, K. F.; Nagai, M.; Wang, H. K.; Itokawa, H.; Lee, K. H. Bioorganic Med. Chem. 2002, 10, 3481–3487.
CrossRef - Mishra, S.; Karmodiya, K.; Surolia, N.; Surolia, A. Bioorganic Med. Chem. 2008, 16, 2894–2902.
CrossRef - Biswas, S. K.; McClure, D.; Jimenez, L. A.; Megson, I. L.; Rahman, I. Antioxid. Redox Signal. 2005, 7, 32–41.
CrossRef - Liang, G.; Li, X.; Chen, L.; Yang, S.; Wu, X.; Studer, E.; Gurley, E.; Hylemon, P. B.; Ye, F.; Li, Y.; Zhou, H. Bioorganic Med. Chem. Lett. 2008, 18, 1525–1529.
CrossRef - Handler, N.; Jaeger, W.; Puschacher, H.; Leisser, K.; Erker, T. Chem. Pharm. Bull. (Tokyo). 2007, 55, 64–71.
CrossRef - Sri Ramya, P. V.; Angapelly, S.; Guntuku, L.; Singh Digwal, C.; Nagendra Babu, B.; Naidu, V. G. M.; Kamal, A. Eur. J. Med. Chem. 2017, 127, 100–114.
CrossRef - Kunwar, A.; Barik, A.; Pandey, R.; Priyadarsini, K. I. Biochim. Biophys. Acta – Gen. Subj. 2006, 1760, 1513–1520.
CrossRef - Khopde, S. M.; Priyadarsini, K. I.; Palit, D. K.; Mukherjee, T. Photochem. Photobiol. 2000, 72, 625-631.
CrossRef - Khadem Sadigh, M.; Zakerhamidi, M. S.; Shamkhali, A. N.; Babaei, E. J. Photochem. Photobiol. A Chem. 2017, 348, 188–198.
CrossRef - Priyadarshini, K. I. J.Photochem. Photobiol. C: Photochem. Rev, 2009, 10, 81-95.
CrossRef - Margar, S. N.; Sekar, N. J. Photochem. Photobiol. A Chem. 2016, 327, 58–70.
CrossRef - Barik, A.; Priyadarsini, K. I. Spectrochim. Acta – Part A Mol. Biomol. Spectrosc. 2013, 105, 267–272.
CrossRef - Zhang, X.; Tian, Y.; Li, Z.; Tian, X.; Liu, H.; Sun, H.; Moore, A.; Ran, C. 2013.
- Nardo, L.; Andreoni, A.; Masson, M.; Haukvik, T.; Tønnesen, H. H. J. Fluoresc. 2011, 21, 627–635.
CrossRef - Margar, S. N.; Rhyman, L.; Ramasami, P.; Sekar, N. Spectrochim. Acta – Part A Mol. Biomol. Spectrosc. 2016, 152, 241–251.
CrossRef - Caselli, M.; Ferrari, E.; Imbriano, C.; Pignedoli, F.; Saladini, M.; Ponterini, G. J. Photochem. Photobiol. A Chem. 2010, 210, 115–124.
CrossRef - Evale, B. G.; Hanagodimath, S. M.; Khan, I. A.; Kulkarni, M. V. Spectrochim. Acta – Part A Mol. Biomol. Spectrosc. 2009, 73, 694–700.
CrossRef - Liang G. Bioorganic Med. Chem. Lett.. 2008, 18, 1525-1529.
CrossRef - Bilot, I.; Kawski, A.; Naturforsch, Z. 1962, 17a, 621-627.
- Kawski, A.; Bojarski, P. Spectrochim. Acta – Part A Mol. Biomol. Spectrosc. 2011, 82, 527–528.
CrossRef - Suppan, P. Chem. Phys. Lett. 1983, 94, 272–275.
CrossRef - Kawski, A.; in: J.F. Rabek(Ed.), Progress in Photochemistry and Photophysics, 5, CRC Press, Boca Raton, USA 1992, pp, 1-47.
- Akshaya, K. B.; Varghese, A.; Lobo, P. L.; Kumari, R.; George, L. J. Mol. Liq. 2016, 224, 247–254.
CrossRef - Kumari, R.; Varghese, A.; George, L. J. Lumin. 2016, 179, 518–526.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


