Theoretical Spectroscopic and Second Harmonic Generations Studies of 5–Fluoro-2-Methyl Benzonitrile
A. Ajay Praveen Kumar1, 2 and R. Ganapathi Raman1, 2
1Department of Physics, Noorul Islam Centre for Higher Education, Kumaracoil - 629 180, Kanyakumari Dist, Tamilnadu, India.
2Nano Computational Laboratory, Department of Nano Technology, Noorul Islam Centre for Higher Education, Kumaracoil - 629 180, Kanyakumari Dist, Tamilnadu, India.
Corresponding Author E-mail: ajaytenoul@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/330534
5-fluoro-2-methylbenzonitrile (5F2MLBN), a novel molecule has been reported after mutually experimental and theoretical approaches on molecular atomic structure, vibrational spectra, non-linear optics (NLO) properties. The FT-IR (400–4000 cm-1) and FT-Raman spectra (50-3500 cm-1) of 5F2MLBN were recorded. The density functional HF methods with three higher basis sets were determined the molecular geometry, harmonic vibrational frequencies and bonding features of 5F2MLBN in the ground state. Finally, spectra of the title compound have good complement while compared with the calculation results were applied to simulate infrared and Raman spectra.
KEYWORDS:5F2MLBN; DFT; VEDA; TED
Download this article as:| Copy the following to cite this article: Kumar A. A. P,Raman R. G. Theoretical Spectroscopic and Second Harmonic Generations Studies of 5–Fluoro-2-Methyl Benzonitrile. Orient J Chem 2017;33(5). |
| Copy the following to cite this URL: Kumar A. A. P,Raman R. G. Theoretical Spectroscopic and Second Harmonic Generations Studies of 5–Fluoro-2-Methyl Benzonitrile. Orient J Chem 2017;33(5). Available from: http://www.orientjchem.org/?p=39176 |
Introduction
Benzonitrile is the child molecule to the parent molecule ammonia to toluene, which had the enormous changing with the consequence of the particular ratio [1]. The fragrances, cosmetics, steroid, aromatic alcohols for color removers, solvents for fatty acids, hydrocarbons and oils are used with benzonitrile as a chemical broker. In recent researchers used Benzonitrile as most excellent solvent and versatile precursor chemical intermediate in many derivatives. Benzonitrile is a cyano group Benzonitrile is a colorless, almond-like odor salt, sharp taste [2] and boiling point at 190.7ºC at 760 mm Hg [3]. Benzonitrile compounds had high toxic and irritating product. World Wide Organization set the range of benzonitrile while using the daily usage products. Toxicity effect used for evaluated in prokaryotic and eukaryotic systems. Which evaluation provided the sharp detail and carcinogenicity, chronic toxicity, and mutagenicity [4]. Anisole (methylbenze) is another form of benzonitrile as it allows for dipolar and dispersion interactions while its capability to be involved in stronger interactions, such as hydrogen bonds, is limited to the role of proton acceptor. In analytical chemistry, the ortho- substituted derivatives of fluro are used as reagents also identified the anesthetic problems and brain diseases are cured through these drugs [5].
Benzoic acid reacts with lead thiocyanates, Benzonitrile phenyl cyanic compound will produced by heating [6]. The pH level of the food will thaw out the effectiveness of benzoic acid. Acidic food, beverages, pickles and acidified foods are preserved with benzoic acid. In the field of medicine, these acid has used as an antibacterial and excellent painkiller in the near the beginning of 20th century [7]. Benzoic acids used for the production of Whitefield’s ointment, tincture and Friar’s balsam [8].
The benzene ring in 5–Fluoro-2-Methyl Benzonitrile (5F2MLBN) has three substituents such that the fluro and methyl groups are at meta and ortho position respectively with respect to the cyano group. The methyl and cyano groups are generally referred to as electron donating substituents in aromatic ring systems. The joining of cyano group in benzonitrile and the aromatic ring creates the migration of the nitrogen atom.
Associations of molecular structural design, nonlinear retort and hyperpolarizability were finding from the vibrational spectra studies of the molecules. The structure of the particle has been leaded by the spectra in juxtaposition through quantum chemical computations. For the spectral approaches unchangeable due to different theoretical methods and series of semi empirical to DFT approaches because every method had its inbuilt capacity [9]. The DFT has been accepted as a popular post-HF approach for the computation of molecular structure, vibrational frequencies and energies of molecule by the ab initio quantum chemistry community [10]. The present letter 5–Fluoro-2-Methyl Benzonitrile (5F2MLBN) have inspected both observed and calculations method. The literature review concluded that, there are no publications of the title compound 5F2MLBN using the theoretical methods. From the equilibrium molecular structure, parameters and vibrational assignments help to determine the results of the interior properties of the molecules. Hyperpolarizability parameters are predicted using the DFT methods using the hybrid basis sets.
Experimental Details
The spectroscopic analytical purpose, The Fourier-transform infrared (FTIR) and The Fourier-transform Raman (FT Raman) spectra have been recorded at normal temperature in the region 400-4000cm-1 and 50-3500cm-1 respectively. Infrared spectrum recorded using 8400S Bruker, Alpha T, and Germany infrared spectrophotometer with scanning speed of 30 cm-1 min-1the spectra are traced. FT Raman spectrum has been recorded using 1064nm line of Nd: YAG laser as excitation wavelength on an EZRaman, Enwaveoptronics, and USA IFS 66 V spectrometer. All spectra recorded out at our campus of the Nanotechnology Department. The spectra are exposed in Fig.2 & Fig.3.
Computational Details
The Gaussian 09W software package has been used for predicting the whole vibrational assignments and optimized geometrical parameters of 5F2MLBN with the original version [11]. The complete geometry parameter has been derived from Becke-Lee-Yang-Parr hybrid (B3LYP) is the three parameter functional by way of applying ab-initio B3LYP hybrid method [12] on Intel Core i3 3.3GHz processor personal computer. The vibrational wavenumbers corrected by introducing the scaling factor. The Scaling factor values are 0.9556 and 0.9959 at (d,p) and cc-pvdz respectively [13]. The total energy distribution (TED) was calculated and explained using the Scaled quantum mechanical program using VEDA.4.0.Software [14] and vibrational modes also calculated through their TED. An elevated degree of precision at the customarily considerations along with obtainable linked by the GAUSSVIEW program are made by the molecules vibrational frequency assignments. From the second derivatives of the energy has calculated the FT-IR, Raman frequencies are computing and intensities plots are done by the Gauss sum program [15]. The non-linear optical (NLO) assets such as dipole moment (µ), polarizability (α), anisotropy of polarizability (α0), and first hyperpolarizability (β0) calculated to understand the frequency doubling the SHG behavior of 5F2MLBN are computed and Thermo dynamical parameters are resolute by Gaussian09 DFT.
Results and Discussion
Molecular Geometry Optimizations
5F2MLBN compound optimized structure has been illustrated (Fig. 1). The stable minimum energy calculated by B3LYP functional with the standard three basis sets (Table 2.1). The predicted atomic lengths, atomic angles and torsion angle or dihedral angles from the above method are tabulated in Table. 2. There is no data on the optimized structure and equilibrium parameters of 5F2MBN do not exist in earlier reports through the literature survey. The microwave data are slightly small while compare with theoretical value of the optimized atomic lengths be in the right places to isolated title compound in gaseous phase. The predicted geometrical parameters of the two different basis sets are almost similar. The theoretical values had good agreement with the microwave data. Benzene ring had six carbon atoms and hydrogen atoms which carbons atoms had same lengths and angles and hydrogen atoms had some changes. The molecule have modify in the different chemical and physical assets because of the hydrogen in benzene ring which acts as the trepidation of the valence electron distribution. The recent molecule interactions to the substituents are indicating the benzene ring angular changes [16].
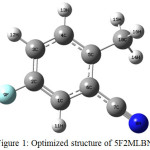 |
Figure 1: Optimized structure of 5F2MLBN. Click here to View figure |
The molecule has seven carbons bonds, eight C-C, six C–H, one C–N and C-F bonds. There is no crystal structure for the 5F2MLBN,benzene ring appears a small hazy with carbon to carbon atom bond length next to the substitutions place (≈1.40Å) longer than the carbon bond (C2-F9) in the middle of the ring substituted in the fluro (≈1.35Å). The distorted elucidates that the substituted on the ring can affect the hybridization and bond lengths of the carbon atoms. The bond length of C-C increase while compare with benzene ring this shows the angle between substituted atoms has little modified hexagonal structure of the angles. From that result, it is found that the benzene ring has almost the same length of the all carbons bond and deviation should be around 0.005 Å it only corresponds with the earlier reports. The length of the C≡N bond had 1.15Å is computed by all basic sets and 1.15 Å is observed for the benzonitrile in early results. All results had good correlation and the minimum deviation level is 0.02Å, whose result is shorter than the single bond carbon and fluro group as valued is 1.35 Å for three basis sets. At last, the length of the carbon fluro bond is calculated and merged with the observed value [17]. The angles between the bonds have been predicted by an atom that has tae corresponding results while comparing with the microwave data.
Table 1: Universal minimizations energy of 5F2MLBN.
| Basic sets | Energy(a.u) |
| B3LYP/6-311++G(d,p) (A) | -463.1727458 |
| B3LYP/cc-pvdz (B) | -463.1727459 |
| B3LYP/Aug-cc-pvdz (C) | -463.0990167 |
Vibrational Spectra
There are 42 vibrational assignments are assigns for the 5F2MLBN molecule. In the 42 assignments C1 point group symmetry. Every vibration intended for N atomic molecules had 2N-3 and N-3 is both bending. As a result 29 in-planes (β) and 13 out of plane (δ) bending are identified for 5F2 MLBN 28 of all 42 vibrations.
Table 2: Equilibrium parameters of 5F2MLBN.
|
Bond Length(Å) |
A |
B |
C |
M.D |
Dihedral Angle(°) |
A |
B |
C |
|
|
C1-C2 |
1.36 |
1.36 |
1.4 |
1.4 |
C6-C1-C2-C3 |
0 |
0 |
-0.003 |
|
|
C1-C6 |
1.54 |
1.54 |
1.4 |
1.4 |
C6-C1-C2-F9 |
180 |
180 |
-180 |
|
|
C1-H11 |
1.07 |
1.07 |
1.07 |
H11-C1-C2-C3 |
180 |
180 |
180 |
||
|
C2-C3 |
1.54 |
1.54 |
1.4 |
1.39 |
H11-C1-C2-F9 |
0 |
0 |
0.0005 |
|
|
C2-F9 |
1.35 |
1.35 |
1.35 |
1.35[18] |
C2-C1-C6-C5 |
0 |
0 |
-0.008 |
|
|
C3-C4 |
1.36 |
1.36 |
1.4 |
1.39 |
C2-C1-C6-C7 |
180 |
180 |
180 |
|
|
C3-H12 |
1.07 |
1.07 |
1.07 |
H11-C1-C6-C5 |
180 |
180 |
180 |
||
|
C4-C5 |
1.54 |
1.54 |
1.4 |
1.39 |
H11-C1-C6-C7 |
0 |
0 |
-0.0076 |
|
|
C4-H13 |
1.07 |
1.07 |
1.07 |
C1-C2-C3-C4 |
0 |
0 |
0.004 |
||
|
C5-C6 |
1.36 |
1.36 |
1.4 |
1.39 |
C1-C2-C3-H12 |
180 |
180 |
-180 |
|
|
C5-C10 |
1.54 |
1.54 |
1.54 |
F9-C2-C3-C4 |
180 |
180 |
180 |
||
|
C6-C7 |
1.54 |
1.54 |
1.4 |
F9-C2-C3-H12 |
0 |
0 |
0.002 |
||
|
C7-N8 |
1.15 |
1.15 |
1.15 |
1.15 |
C2-C3-C4-C5 |
0 |
0 |
0.0055 |
|
|
C10-H14 |
1.07 |
1.07 |
1.07 |
C2-C3-C4-H13 |
180 |
180 |
-180 |
||
|
C10-H15 |
1.07 |
1.07 |
1.07 |
H12-C3-C4-C5 |
180 |
180 |
-180 |
||
|
C10-H16 |
1.07 |
1.07 |
1.07 |
H12-C3-C4-H13 |
0 |
0 |
0.004 |
||
|
Bond Angle (°) |
C3-C4-C5-C6 |
0 |
0 |
-0.02 |
|||||
|
C2-C1-C6 |
120 |
120 |
120 |
C3-C4-C5-C10 |
180 |
180 |
180 |
||
|
C2-C1-H11 |
120 |
120 |
120 |
H13-C4-C5-C6 |
180 |
180 |
180 |
||
|
C6-C1-H11 |
120 |
120 |
120 |
H13-C4-C5-C10 |
0 |
0 |
-0.02 |
||
|
C1-C2-C3 |
120 |
120 |
120 |
122.8[18] |
C4-C5-C6-C1 |
0 |
0 |
0.02 |
|
|
C1-C2-F9 |
120 |
120 |
120 |
118.7[18] |
C4-C5-C6-C7 |
180 |
180 |
-180 |
|
|
C3-C2-F9 |
120 |
120 |
120 |
118.5[18] |
C10-C5-C6-C1 |
180 |
180 |
-180 |
|
|
C2-C3-C4 |
120 |
120 |
120 |
119.9 |
C10-C5-C6-C7 |
0 |
0 |
0.02 |
|
|
C2-C3-H12 |
120 |
120 |
120 |
C4-C5-C10-H14 |
-150 |
-150 |
-90 |
||
|
C4-C3-H12 |
120 |
120 |
120 |
C4-C5-C10-H15 |
-30 |
-30 |
30 |
||
|
C3-C4-C5 |
120 |
120 |
120 |
120.2 |
C4-C5-C10-H16 |
90 |
90 |
150 |
|
|
C3-C4-H13 |
120 |
120 |
120 |
C6-C5-C10-H14 |
30 |
30 |
90 |
||
|
C5-C4-H13 |
120 |
120 |
120 |
C6-C5-C10-H15 |
150 |
150 |
-150 |
||
|
C4-C5-C6 |
120 |
120 |
120 |
120.1 |
C6-C5-C10-H16 |
-90 |
-90 |
-30 |
|
|
C4-C5-C10 |
120 |
120 |
120 |
||||||
|
C6-C5-C10 |
120 |
120 |
120 |
||||||
|
C1-C6-C5 |
120 |
120 |
120 |
120.2 |
|||||
|
C1-C6-C7 |
120 |
120 |
120 |
||||||
|
C5-C6-C7 |
120 |
120 |
120 |
||||||
|
C5-C10-H14 |
110 |
110 |
109 |
||||||
|
C5-C10-H15 |
110 |
110 |
109 |
||||||
|
C5-C10-H16 |
110 |
110 |
109 |
A- B3LYP/6-311++G(d,p) |
|||||
|
H14-C10-H15 |
110 |
110 |
109 |
B- B3LYP/cc-pvdz |
|||||
|
H14-C10-H16 |
110 |
110 |
109 |
C- B3LYP/Aug-cc-pvdz |
|||||
|
H15-C10-H16 |
110 |
110 |
109 |
M.D – Microwave Data (benzonitrile) |
|||||
In the novel compound of 5F2MLBN are premeditated the harmonic vibrational frequencies at B3LYP level by various basis sets have specified. Experimental data and the portrayals alarming the mission also have designated in Table.3 gives the coherent basis for the assignments and shows that the molecules assignments as well as IR & Raman intensities of the title molecule. In Figs.2 and 3 shows the comparisons of experimental and calculated spectra.
Table 3: The observed (FT-IR, FT-Raman) and calculated frequencies (cm-1), IR intensity (km mol-1), Raman intensity (Å4 amu-1) and probable assignments of 5F2MLBN.
|
Normal Modes |
Experimental (cm-1) |
Scaled Wavenumbers(cm-1) |
Raman Activity |
Assignments with TED (%) |
||||
|
Mode |
Label |
FT-IR |
FT-Raman |
A |
B |
IR Intensity(Km/mol) |
||
|
1 |
A |
79 |
80 |
0.13 |
0.4102 |
|||
|
2 |
A |
130 |
130 |
0.21 |
0.7025 |
τCCCC(69)+τCCCN(12) |
||
|
3 |
A |
144 |
143 |
143 |
0.23 |
3.9458 |
βCCC(48)+βCCN(44) |
|
|
4 |
A |
154 |
154 |
0.25 |
0.2942 |
τCCCC(51)+τCCCF(27) |
||
|
5 |
A |
304 |
304 |
0.48 |
0.426 |
βCCC(53)+βCCF(20) |
||
|
6 |
A |
330 |
337 |
337 |
0.54 |
2.0035 |
τCCCF(45)+τCCCC(37) |
|
|
7 |
A |
403 |
403 |
0.64 |
1.4563 |
τCCCC(48)+τCCCN(34) |
||
|
8 |
A |
415 |
415 |
0.66 |
0.5394 |
βCCF(53)+βCCN(15)+βCCC(13) |
||
|
9 |
A |
434 |
446 |
446 |
0.71 |
5.7741 |
βCCC(58)+νCC(10) |
|
|
10 |
A |
484 |
492 |
492 |
0.78 |
0.474 |
τCCCC(67)+τCCCN(18) |
|
|
11 |
A |
531 |
498 |
498 |
0.79 |
9.3586 |
νCC(30)+βCCC(27)+νFC(10) |
|
|
12 |
A |
578 |
602 |
602 |
0.96 |
1.2148 |
βCCC(34)+βCCN(30) |
|
|
13 |
A |
666 |
627 |
627 |
1 |
1.9935 |
τCCCC(60)+τCCCN(17)+τCCCF(15) |
|
|
14 |
A |
701 |
708 |
720 |
720 |
1.15 |
0.3561 |
τCCCC(78) |
|
15 |
A |
723 |
723 |
1.15 |
7.6416 |
βCCC(46)+νCC(15)+νFC(10) |
||
|
16 |
A |
750 |
758 |
764 |
764 |
1.22 |
16.5348 |
βCCC(39)+νCC(28) |
|
17 |
A |
819 |
838 |
837 |
1.33 |
0.0978 |
τHCCF(50)+δCCCH(37) |
|
|
18 |
A |
887 |
898 |
898 |
1.43 |
0.0739 |
δCCCH(91) |
|
|
19 |
A |
937 |
944 |
955 |
955 |
1.52 |
14.6849 |
νCC(22)+νFC(18)+βCCC(14) |
|
20 |
A |
965 |
965 |
1.54 |
0.072 |
δCCCH(49)+τHCCF(40) |
||
|
21 |
A |
1020 |
1020 |
1.63 |
2.5262 |
βHCC(29)+δCHCH(28)+νCC(11) |
||
|
22 |
A |
1081 |
1062 |
1062 |
1.69 |
0.134 |
βHCC(58)+δCHCH(28) |
|
|
23 |
A |
1117 |
1117 |
1.78 |
2.1453 |
βHCC(25)+νCC(14)+βCCC(11) |
||
|
24 |
A |
1142 |
1170 |
1170 |
1.86 |
1.8219 |
βHCC(56)+νCC(20) |
|
|
25 |
A |
1209 |
1209 |
1.93 |
6.4833 |
νCC(40)+βHCC(25)+βCCC(10) |
||
|
26 |
A |
1247 |
1278 |
1272 |
1272 |
2.03 |
60.2073 |
νFC(21)+νCC(10) |
|
27 |
A |
1292 |
1291 |
2.06 |
2.5717 |
βHCC(54) |
||
|
28 |
A |
1390 |
1325 |
1325 |
2.11 |
6.1959 |
νCC(56) |
|
|
29 |
A |
1423 |
1423 |
2.27 |
11.9581 |
βHCH(49)+βHCC(26)+δCHCH(22) |
||
|
30 |
A |
1459 |
1434 |
1434 |
2.29 |
2.551 |
νCC(39)+ βHCC(13) |
|
|
31 |
A |
1484 |
1484 |
2.36 |
8.6356 |
δCHCH(65)+βHCC(32) |
||
|
32 |
A |
1495 |
1496 |
2.38 |
4.7715 |
βHCH(41)+βHCC(23)+δCHCH(23) |
||
|
33 |
A |
1511 |
1521 |
1521 |
2.42 |
4.2765 |
βHCC(33)+βCCC(22) |
|
|
34 |
A |
1617 |
1617 |
2.58 |
16.1349 |
νCC(39)+ βCCC(11) |
||
|
35 |
A |
1649 |
1664 |
1648 |
1648 |
2.63 |
60.4469 |
νCC(66) |
|
36 |
A |
2222 |
2334 |
2230 |
2230 |
3.71 |
390.9454 |
νNC(89)+ νCC(11) |
|
37 |
A |
3064 |
3033 |
3033 |
4.83 |
195.3965 |
νCH(99) |
|
|
38 |
A |
3065 |
3084 |
3083 |
4.91 |
63.8787 |
νCH(98) |
|
|
39 |
A |
3116 |
3116 |
4.97 |
55.0693 |
νCH(81) |
||
|
40 |
A |
3179 |
3179 |
5.07 |
73.9126 |
νCH(91) |
||
|
41 |
A |
3203 |
3203 |
5.1 |
162.9662 |
νCH(91) |
||
|
42 |
A |
3212 |
3212 |
5.12 |
85.3282 |
νCH(99) |
||
|
ν- stretching; β- in plane bending; δ- Out-of-plane bending; τ-torsion; TED-Total Energy Distribution. |
||||||||
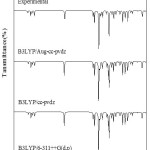 |
Figure 2: Experimental and theoretical DFT/B3LYP FT-IR of 5F2MLBN. Click here to View figure |
 |
Figure 3: Experimental and theoretical DFT/B3LYP FTR of 5F2MLBN. Click here to View figure |
CH Vibrations
Four kinds of CH moieties are calculated in the title compound 5F2MLBN. In this CH moieties have been listed, six CH stretching, in plane as well as torsion vibrations. The scenery of the substituent does not align because of these stretching vibrations. The stretching has most feasible frequency between the regions at 3100-3000cm-1 [18]. The calculated stretching vibrations of the title compound region, in between 3202-3005cm-1. There is no experimental frequencies have been observed. The calculated CH in-plane bending vibrations of 5F2MLBN occurs in the region of 1550-1000cm-1 and observed FTIR at 1511, 1459, 1142, 1051 cm-1 [19]. The CH out-of-plane bending occurs at the region 1100-800cm-1 generally. The calculated out-of-plane bending vibrations of 5F2MLBN are found at 819, 887, 1081 cm-1. Observed FTIR and FTR spectra occurred in the region 876 and 908cm-1 correspondingly. In this case, the in-plane and out-of-plane bending has been merged with the torsion vibrations. Calculate and observed values good agreement with each other.
C≡N Vibrations
In the CN moieties, stretching frequency is the intensively localized one due to the TED for this frequency contains good contribution from that constant value of stretching force. In the benzonitrile molecule, the cyano group (C≡N) vibrational wave number is almost unchanged because of it joined together the phenyl ring as a new substituent [21]. For the aromatic compound which bears a C≡N group attached to the ring, a band of good intensity has been absorbed in the region 2240–2221 cm-1[22] and it is being attributes to C≡N stretching. FL is the electron acceptor groups; it decrease the IR band intensity and increases the wave number value to the higher limit of the characteristic spectral region. CH3 is the electrons donating groups, it increase IR intensity and decrease wave number value. These electron acceptor groups are the main impact for the shifting for C≡N vibrational wave number. FTIR and FTR spectra frequency at 2222 and 2334 cm-1 for C≡N (ν) vibrations found for 5F2MLBN, respectively. The calculated wavenumbers of stretching vibrations at 2230 cm-1 coincide with the FTR value. This is the ideal frequency of the whole molecule which confirmed the CN stretching with 89% assignment. The bending vibrations (β) of the molecule for FTIR spectra spotted at 578 cm-1. These experiment values are well agreement with the calculated frequencies.
C-C and C-C-C Vibrations
Benzonitriles are the substituents cyanogens groups and the benzene ring has almost the same length of the all carbons bond and minimum deviation. The thirteen stretching (mode no. 9,11,15,16,19,21,23,24,26,28,30,34,35,36) carbon vibrations slouch in the region 400-2300 cm-1 and thirteen bending of vibrations slouch above the region 1300 cm–1[23]. From the research, the six similar carbon bonding can produce benzene the same time that ring vibrating the seven C-C stretching modes are scanned through FTIR in the region at 2222, 1649, 1459, 1390, 1247, 1142, 937, 750, 531 cm-1 and the five FTR at 2334, 1664, 1278, 944, 758, 434 cm-1 coincides each other. The recorded spectral values of C-C-C bending modes at 1511, 937, 750, 531 and 944, 758, 434, 144 cm-1 values are equal with predicted numerical.
CCCC Torsional Vibrations
The ring torsions have been assigned in the region at below 800cm-1 which is discussed in present paper referred by earlier reports [24]. The IR and Raman spectra peak observe in the region at 701, 666 cm-1 and 708, 484 cm-1, respectively. These peaks have been calculated at the region at 720, 627, 492, 403, 154, 130 cm-1by B3LYP methods which are well merge with observed values. And these wavenumbers are mixed with CCCN torsions in the investigated molecules.
Optical Property
The hyperpolarizability is contributed by the structure, bonding and vibrational of the molecule. The dipole moment (µ) and hyperpolarizability of the benzonitrile is high. In the recent molecule has been calculated the enhanced hyperpolarizability value which is due to the substituent of the benzonitrile. The bond and vibrational results are confirmed the envelope to the hyperpolarizability enlargement of the 5F2MLBN molecule. In the fields such as telephoning, signal transferring and fiber optic cables, NLO enhance the functions for the developing technologies like frequency modulation, optical changing, optical controlling and optical logical circuits [25]. The first hyperpolarizability (β) polarizability (α) and anisotropy of polarizability [26] ( α) of 5F2MLBN is calculated using DFT with the above basic set and can be evaluated using equations (1) (2) (3) respectively. The Table.4 listed the numerical values of above mentioned parameters. The electric is the task for the energy of the scheme because an electrical meadow exists in the molecule. 3 3 3 matrix is used for determined the hyperpolarizability. Kleinman symmetry diminished the 27 mechanisms into 10 mechanisms [27].
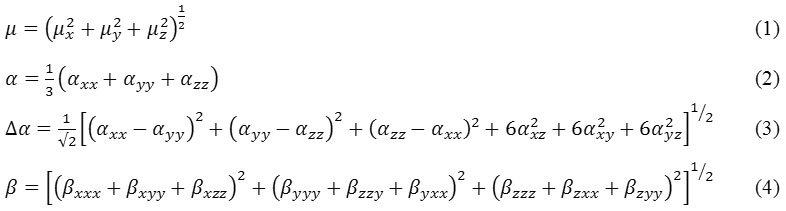
Table 4: Optical properties of 5F2MLBN.
|
Parameters |
B3LYP |
Parameters |
B3LYP |
|
μx |
0.8197256 |
βxxx |
80.156319 |
|
μy |
-0.01 |
βxxy |
-2.4476356 |
|
μz |
1.3473167 |
βxyy |
26.3796534 |
|
μ |
1.577Debye |
βyyy |
-8.36091 |
|
αxx |
101.757788 |
βxxz |
-35.3219987 |
|
αxy |
-0.3604599 |
βxyz |
6.6626508 |
|
αyy |
53.7128032 |
βyyz |
28.5648953 |
|
αxz |
-4.5871788 |
βxzz |
21.4224614 |
|
αyz |
0.28 |
βyzz |
3.8015823 |
|
αzz |
128.7684006 |
βzzz |
-81.8304551 |
|
α0 |
198.393×10-33esu |
β0 |
1345.907×10-33esu |
|
α |
1713.98×10-33esu |
The calculated values of α and β are 1713.98×10−33esu and 1345.907×10−33esu. Ideal molecule urea is used for determined the comparative purpose. The compound has the values 1.577D and 1345.907×10−33esu of μ and β respectively. The μ of compound is approximately 1.15 times greater than that of urea and the β of compound is 3.62 times greater than that of urea (µ and β of urea are 1.3732 Debye and 0.3728 ×10-30esu with the same method). The recent molecule has steady state potential for NLO applications like frequency doubling and communications that is confirmed by the recent research [28].
Conclusion
From the DFT methods by three basis sets, computed the geometrical parameters and optimized structure. The research the experimental FT-IR, FTR spectroscopic studies are investigated first time and explained vibrations assignments with TED% using the B3LYP method. The theoretical spectra are predicted using computational methods and well agreement with experimental results. The μ and β values of the compound were 1.15 and 3.62 times greater than that of urea, respectively. These properties show that the title compound 5F2MLBN had good chemical stability, bioactivity and optics applications are helping the future researchers and innovative thinkers.
References
- Ajaypraveenkumar, A.; Raman, R.G. J. Chem. Pharm. Sci. 2017, 27-41.
- Rastogi, V.K.; Surabhi Singha, Palafox, M.A.; Ramana Rao, G. Indian J. O. Phy. 2010, 84(2), 151-165.
CrossRef - Chaitanya, K. Spectrochim. Acta, Part A. 2012, 86, 159– 173.
CrossRef - Binoy, J.; James, C.; Hubert Joe, L.; Jayakumar, V.S. J. O. mol. Struc., 2006, 784(1), 32–46.
- Sundaraganesan, N.; Ilakiamani, S.; Saleem, H.; Wojciechowski, P.M.; Michalska, D. Spectroc. Acta. 2005, 61A, 2995–3001.
CrossRef - Virendrakumar, Panikar, Y.; Palafox, M.A.; Vats, J.K.; Kostova, I.; Lang, K.; Rastogi, V.K. Indian J. Pure Applied Phys. 2010, 48, 85-94.
- Elanthiraiyan, M.; Jayasudha, B.; Arivazhagan, M. Spectrochim. Acta, Part A. 2015, 134, 543–552.
CrossRef - Saravanan, R.R.; Seshadri, S.; Gunasekaran, S.; Mendoza-Merono, R.; Garcia-Granda, S. Spectrochim. Acta, Part A. 2015, 139, 321–328.
CrossRef - Ventura, C.; M.; Kassab, E.; Buntinx, G.; Poizat, O. Phys. Chem. Chem. Phys. 2000, 2(20), 4682–4689.
- Krishnan, A.R.; Saleem, H.; Subashchandrabose, S.; Sundaraganesan, N.; Sebastain, S. Spectrochim. Acta, Part A. 2011, 78, 582–589.
CrossRef - Sundaraganasan, N.; Elango, G.; Sebastian, S.; Subramani, P. Indian J. Pure Applied Phys. 2009, 47, 481-490.
- Parr, R.R.; Yang, R.G. Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, USA, 1989.
- Ajaypraveenkumar, A.; Raman, R.G. J. Chem. Pharm. Sci. 2017, 10, 1309-1316.
- Murugan, M.; Balachandran, V.; Karnan, M. J. Chem. Pharm. Res. 2012, 4(7), 3400-3413.
- Almosawe, A.J.; Saadon, H.L. Chin. Opt. Lett. 2013, 11 (4), 041902-041906.
CrossRef - Lee, C.; Yang, W.; Parr, R.G. Phys. Rev. B. 1988, 37, 785–789.
CrossRef - Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Chem. Phys. Lett. 1989, 157(3), 200-206.
CrossRef - Huralikoppi, A.M. PhD thesis, Karnataka University Dharrwad, 1995.
- Diwaker, Chidan Kumar, C.S.; Kumar, A.; Chandraju, S.; Fun, H.K.; Quah, C.K. J. Mol. Struct. 2015, 1092, 192-201.
CrossRef - Hubert Joe, I.; Kostova, I.; Ravikumar, C.; Amalanathan, M.; Pinzaru, S.C. J. Ram. Spectrosc. 2009, 40, 1033-1038.
CrossRef - Chen, Y.; Yang, J.; Li, Z.; Li, R.; Ruan, W.; Zhuang, Z.; Zhao, B. Spectrochim. Acta, Part A. 2016, 153, 344-348.
CrossRef - Fleming, I. Frontier Orbitals and Organic Chemical Reactions, Wiley, London, 1976.
- Premkumar, S.; Jawahar, A.; Mathavan, T.; Kumara Dhas, M.; Sathe, V.G.; Benial, A.M.F. Spectrochim. Acta, Part A. 2014, 129, 74–83.
CrossRef - Saravanan, S.P.; Sankar, A.; Parimala, K. J. Mol. Spectrosc. 2017, 1127, 784-795.
- Shajikumar.; Ajaypraveenkumar, A.; Raman, R.G. J. Chem. Pharm. Sci. 2017, 2063-2072.
- Eryılmaz, S.; Gul, M.; Inkaya, E.; Idil, O.; Ozdemir, N. J. Mol. Struct. 2016, 1122, 219-233.
CrossRef - Kleinman, D.A. Phys. Rev. 1962, 126(6), 1977-1979.
CrossRef - Ajay praveen kumar, A.; Raman, R.G. J. Chem. Pharm. Sci. 2017, 1-9.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


