Oxidation of Hydroxylamine by Waugh Type POM, Enneamolybdomanganate(IV) Ion: A Kinetic and Mechanistic Study
J. N. Barge and G. S. Gokavi
Department of Chemistry, Shivaji University, Kolhapur 416 004, Maharashtra, India.
Corresponding Author E-mail: gsgokavi52@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/330554
The reaction between hydroxyl amine and Waugh-type polyoxometalate, enneamolybdomanganate(IV), was investigated under pseudo-first-order condition in aqueous perchloric acid. The product of the reaction was nitrous oxide as determined by studying the stoichiometry which requires one mol of oxidant per mole of hydroxylamine. The rate of reaction increases as hydrogen ion and oxidant, enneamolybdomanganate(IV), concentrations increase, while it remains unchanged as the hydroxylamine concentration is altered. The oxidant exists in both protonated and unprotonated form under the reaction conditions and both are active. The hydroxyl amine remains in the protonated form and reacts with the active forms of the oxidant with different rate constants. The proposed mechanism, in accordance with the kinetic results obtained, involves reaction between hydroxyl amine and oxidant in a slow step. The rate law derived as per the proposed mechanism is also verified kinetically. Change in ionic strength and solvent polarity did not affect the rate of the reaction while activation parameters were calculated by studying the reaction at different temperatures.
KEYWORDS:Polyoxometalate Oxidation; Reaction Mechanism; Enneamolybdomanganate; Hydroxylamine
Download this article as:| Copy the following to cite this article: Barge J. N, Gokavi G. S. Oxidation of Hydroxylamine by Waugh Type POM, Enneamolybdomanganate(IV) Ion: A Kinetic and Mechanistic Study. Orient J Chem 2017;33(5). |
| Copy the following to cite this URL: Barge J. N, Gokavi G. S. Oxidation of Hydroxylamine by Waugh Type POM, Enneamolybdomanganate(IV) Ion: A Kinetic and Mechanistic Study. Orient J Chem 2017;33(5). Available from: http://www.orientjchem.org/?p=37558 |
Introduction
Hydroxylamine is an intermediate between nitrous oxide and ammonia, it can be oxidized to nitrous oxide and reduced to ammonia1. Its redox chemistry also has biological importance as in nitrification it is oxidized by the enzyme hydroxylamine oxidoreductase(HAO) obtained from nitrosomonas europaea bacteria to nitrite2-5. Hydroxylamine can be obtained back through reduction of nitrite by another enzyme nitrite reductase6. Therefore, hydroxylamine is an important intermediate if the oxidation reduction transformation of biological nitrification reaction. The primary mechanism of oxidation of hydroxylamine to nitrous oxide7-9 by the HAO is the reduction of its FeIII containing redox centre to FeII. Hydroxylamine is also used in organic synthesis10 in combination with oxidant like iodate for conversion of alcohols to corresponding carbonyl compounds selectively. The mechanism proposed for oxidation of alcohols involve formation of a nitrosated intermediate as a result of formation of nitric oxide. Substituted hydroxylamine are also used as a ligand for metals1 and metal oxides11 for formation of oxime complexes. These oxime complexes are used as catalysts for oxidation of alcohols, sulphides, amines and olefins to epoxide11.
Polyoxometalates(POM) are the stable anionic metal oxygen clusters with or without a heteroatom either at the centre or at the surface. These POMs with discrete structure resemble enzymes in their activities specifically the oxidative transformations12-14. The properties of the hetero atom either at the centre in POM or at the surface of a lacunary POM can be altered so that they can be used for specific catalysis. The POMs can be utilized as the electron transfer, oxygen transfer or radical pathways as well as solid acids13, 15-16. Due to such versatility of POM they are considered as model metalloenzymes in biologically analogous oxidations. Therefore, interaction between the redox active metal centres in a POM with hydroxylamine another biologically important simple molecule in nitrification reaction is of mechanistic interest.
It has been also noticed that the MnIII and FeIII substituted POMs have shown good catalytic activity in transferring oxygen from idosobenzene to either alkene or alkane17-18. The activity is anaologous to metallophorphyrins and the MnIII-POM was found to be more active than that of the FeIII-POM. Therefore, we have considered a Waugh type POM, enneamolybdomanganate(IV), as an oxidant to understand its reactivity towards hydroxylamine. The oxidant used in the present study is composed of central MnIVO6 unit surrounded by nine MoO6 octahedra. The redox potential of the MnIV/MnII couple19 in the POM is about 1.035 V which is considerably lower that the value20 of 1.51 V for free couple thus making it as a mild oxidizing agent. Since the oxidation of hydroxylamine by HAO involves FeIII/FeII redox couple in its mechanism we investigated its oxidation by enneamolybdomanganate(IV) which also involve MnIV/MnII redox couple21.
Experimental
Materials
Reagent grade chemicals and double-distilled water was used to prepare the solutions required. A known amount of hydroxylamine hydrochloride (SD Fine) was dissolved in double-distilled water every day to obtain its stock solution. A known volume of perchloric acid was diluted with double-distilled water to get its solution. The ammonium salt of MnIV complex, (NH4)6[MnIVMo9O32] was synthesized according to reported method22. The synthesized polyoxometalate was characterized by FTIR and AAS to understand the structure and formula of the compound23-24. The reaction was followed by measuring the absorbance of the reaction mixture using Elico SL-177 spectrophotometer.
Kinetic Measurements
While studying the reaction, pseudo-first order conditions were maintained in which [NH2OH] was in large excess over [oxidant] in a thermostat kept at 25 ± 0.1°C. To initiate the reaction, known volumes of the earlier thermostated solutions of NH2 OH, (NH4)6 [MnIV Mo9O32] and perchloric acid were mixed. The advancement of the reaction was monitored spectrophotometrically at 468 nm (ε = 360 ± 2 dm3 mol−1 cm−1) by measuring the change in absorbance of oxidant upto 80 % completion of the reaction. From the slope of plot of log [oxidant] versus time the pseudo-first order rate constants were calculated which were found to be reproducible within ±5%. Reaction mixtures containing constant [MnIVMo9O32]6− of 2.0×10−3 and [NH2OH] from 0.5 x 10-4 to 2.5×10-4 mol dm−3 were prepared and analysed for unreacted oxidant after 24 h to determine the stoichiometry. For each mol of NH2OH in the reaction mixture one mole of oxidant was consumed. Therefore, the reaction stoichiometry is 1;1 mol of NH2OH per mole of oxidant which indicate nitrous oxide as the product according to the equation 1. A similar stoichiometry for the oxidation of hydroxylamine by Fe(III), [CoW12O40]5-, AuCl4– and peroxydisulphate 25-28 was reported
H6[MnIVMo9O32] + NH2OH → H8[MnIIMo9O32] + ½ N2O + H+ (1)
Results and Discussion
The formation of free radical in a reaction is examined by studying the reaction by adding acrylonitrile which is free radical scavenger. The addition of acrylonitrile between 10 to 40 % V/V to the reaction mixture did not alter the rate constant nor there was a precipitation due to the polymerization of acrylonitrile. Therefore, from the absence of precipitate due to polymerization and no change in the rate constant it can be concluded that formation of free radical intervention is not occurring in the reaction. The effect of added molybdate, MnSO4 and the ionic strength also did not affect the rate of the reaction. In order to find the order in [oxidant] a plots of log [oxidant] against time for the [oxidant] between 0.36 x10-3 to 3.6 x 10-3 mol dm-3 were examined and all the plots were linear upto more than 93 % of the reaction and the pseudo-first-order rate constants are also constant (Table 1). The constancy in rate constants as [oxidant] varies and the linearity of pseudo-first-order plots show that the order in [oxidant] is one. Similarly the constancy of the pseudo-first-order rate constants (Table 1) as [NH2OH] changes from 5.0 x 10-3 to 5.0×10-2 mol dm-3 at a constant [oxidant] of 1.5 x 10-3 mol dm-3 shows the order of one in [NH2OH]. The concentration of perchloric acid was varied between 0.05 and 0.5 mol dm-3 to understand the dependence of reaction on the [H+]. The reaction is catalysed (Table 2) by [H+] and the order in [H+] was found to be less than one (0.5). Therefore, a graph of pseudo-first-order rate constants, kobs, against [H+] was plotted and found to be linear (Figure 1) with an intercept indicating that the reaction follows [H+] dependent and independent paths. The variation in ionic strength and solvent polarity were studied at 25°C by sodium perchlorate and acetonitrile. Variation in both ionic strength and solvent polarity did not affect the reaction.
![Figure 1: Plot of kobs against [H+] at different temperatures.](http://www.orientjchem.org/wp-content/uploads/2017/09/Vol33No5_Oxi_Bar_fig1-150x150.jpg) |
Figure 1: Plot of kobs against [H+] at different temperatures. |
Table 1: Effect of concentration of enneamolybdomanganate(IV) and hydroxylamine on the rate of reaction at 25oC.
|
102[NH2OH] mol dm-3 |
103[MnIVMo9O32]6- mol dm-3 |
104 kobs s-1 |
|
1.0 |
0.36 |
1.01 |
|
1.0 |
1.1 |
2.96 |
|
1.0 |
1.5 |
3.80 |
|
1.0 |
2.7 |
6.91 |
|
1.0 |
3.6 |
9.98 |
|
0.5 |
1.5 |
3.80 |
|
1.0 |
1.5 |
3.83 |
|
1.5 |
1.5 |
3.90 |
|
2.0 |
1.5 |
3.91 |
|
5.0 |
1.5 |
3.85 |
[HClO4] = 0.2 mol dm-3, I = 0.3 mol dm-3.
Table 2: Effect of perchloric acid concentration on the rate of reaction.
|
Temperature K |
104 kobs s-1 at |
||||
|
[H+] = 0.05 mol dm-3 |
[H+] = 0.1 mol dm-3 |
[H+] = 0.2 mol dm-3 |
[H+] = 0.4 mol dm-3 |
[H+] = 0.5 mol dm-3 |
|
|
288 |
0.77 |
1.15 |
1.92 |
2.30 |
2.69 |
|
293 |
1.15 |
1.54 |
2.30 |
3.45 |
4.22 |
|
298 |
1.90 |
2.50 |
3.68 |
5.24 |
6.03 |
|
303 |
2.69 |
3.10 |
4.61 |
7.14 |
8.91 |
|
313 |
4.14 |
5.13 |
7.23 |
10.4 |
12.2 |
103[MnIVMo9O32]6- = 1.5 mol dm-3, 102 [NH2OH] = 1.0 mol dm-3, I = 0.6 mol dm-3.
The UV-VIS spectrum of enneamolybdomanganate(IV) anion shows a charge transfer absorption band at 468 nm which is characteristic of the MnIV ion29 due to A2g → 4T2g transition. The UV-VIS spectrum of this anion shows two isobestic points due to the protonation22. The absorption band at 468 nm shifts when there is change in the environment around the MnIV ion29. Therefore, when the reaction mixture containing both the reactants is examined spectrophotometrically, the isosbestic points at 435 and 522 nm remain unchanged as well as the intensity of both the peaks decrease. If hydroxylamine interacts with the oxygen atoms of the MnO6 octahedra of the oxidant then the band at 468 nm would have been reappeared. Instead, the existence of isosbestic point in the UV-VIS spectrum of the reaction mixture and decrease in intensity is due to absence of interaction of hydroxylamine through oxygen atoms bound to MnO6 octahedra and also absence of formation of a strong complex between the reactants.
The coordination chemistry of hydroxylamine rarely studied except as a transition state in its transition metal or metal catalysed oxidations30. Considering the probable binding sites of hydroxylamine would be either the N or the O atom various metal complexes of N,N-bis(2-{pyrid-2-ylethyl})hydroxylamine were synthesized1. The complexation was found to occur through neutral amine group of hydroxylamine as a binding site and these complexes were also reduced by the metal ions. Therefore, the binding site in the hydroxylamine is the neutral, unprotonated, amine group. The present study was carried out in acidic solutions and since the pK of amine group30 is 5.9 it will be in the protonated form. Therefore, the complex formation through amine group in the present study is not feasible. Since, the hydroxylamine is completely protonated at all hydrogen ion concentration used in the present study, the effect of hydrogen ion concentration on the rate will not be due to the change in the reactive species of the reductant. On the other hand, the oxidant, [MnMo9O32]6- , exists in the protonated, H6[MnMo9O32], form as evidenced by its UV-VIS spectra in acidic conditions and further dissociates to give H5[MnMo9O32]–. Therefore, under the present reaction conditions, both H6[MnMo9O32] and H5[MnMo9O32]– are present in solution. The reaction follow a two term rate law with [H+] dependent and independent indicating (Fig 1) both H6 [MnMo9O32] and H5 [MnMo9O32]– are reactive species of the oxidant. Therefore, the mechanism considering both H6[MnMo9O32] and H5[MnMo9O32]– as active oxidant species and protonated hydroxylamine, NH3OH+, as the active reductant species can be represented as in Scheme I. According to Scheme I the rate law can be derived as follows.
The expression derived for the pseudo-first-order rate constant, kobs, as in equation 11 satisfy all the kinetic results obtained. In the expression 11, there is no term containing the concentrations of wither [Mn2+] and [MoO4–] indicating that the polyoxometalate, [MnMo9O32]6-, remain intact before the reaction and after the reaction. Since, the effect of acrylonitrile did not form any precipitate and also did not affect the kobs values it can be concluded that the reaction did not involve a two electron transfer step. The reaction proposed in Scheme I proceeds through two paths one dependent on [H+] and the other independent of [H+]. The [H+] dependent path has the greater rate constant than that of the [H+] independent path as noticed from slope and intercept of Figure 1.The former major path involves a neutral species of the oxidant therefore, the change in ionic strength did not affect the reaction as noticed by studying the reaction at different sodium perchlorate ion concentrations. The rate law 11 can also be verified by plotting kobs against [H+] and which was found to be linear with an intercept (Figure 1) thus justifying the mechanism suggested. The effect of solvent polarity on the reaction was studied by varying the acetonitrile content in the reaction mixture. Since the major path of the reaction contains a neutral oxidant species again, as in case of ionic strength effect, the rate constant remain unaffected even after changing the solvent polarity. The polyoxometalates are outer-sphere oxidants and interaction between the hydroxylamine and [MnMo9O32]6- was also not noticed during the UV-VIS spectrophotometric examination of the reaction mixture. Therefore, the probable transition state in the present reaction is replacement of a water molecule of [MnMo9O32]6- by NH2OH+ ion to form a precursor complex. Such precursor complex is rather loosely bound thus leading to moderate decrease in entropy of activation as noticed in the present study( Table 3). The energy of activation of the [H+] dependent path, in which the active oxidant species is neutral, is higher than that of [H+] independent path, in which it is in the anionic form due to dissociation. In the latter path the collision between both the reactants is more feasible as they are oppositely charged thus reducing the energy of activation than that in the former path as one of them is neutral which requires comparatively more energy of activation ( Table 3). Since, the hydroxylamine is in the protonated from the formation of precursor complex between two oppositely charged reactants is more favoured with more decrease in activation entropy than when one of them is neutral with less decrease in entropy of activation ( Table 3).
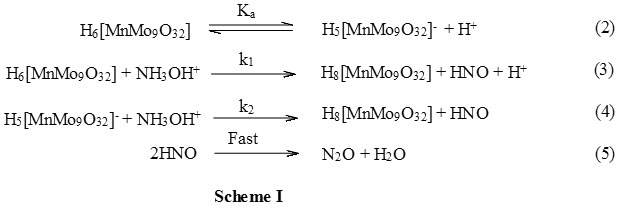

Table 3: Activation parameters for the [H+] dependent and independent term of rate law.
|
Activation parameter |
[H+] dependent term |
[H+] independent term |
|
Ea kJmol-1 |
56.6 + 2 |
45.9 + 2 |
|
DH# kJmol-1 |
42.0 + 2 |
43.7 + 2 |
|
-DS# J K-1mol-1 |
121.4 + 5 |
-172.0 + 5 |
|
DG# kJmol-1 |
78.2 + 2 |
94.9 + 2 |
Conclusions
The reaction between enneamolybdomanganate(IV) and hydroxylamine in aqueous perchloric acid was found occur by direct two-electron transfer step without formation of precursor complex. The hydroxylamine under the experimental conditions remains completely protonated while both protonated and unprotonated forms of enneamolybdomanganate(IV) are the active oxidant species thus making the reaction to be catalysed by hydrogen ion concentration. The rate law consists of two terms containing hydrogen dependent and independent terms. The activation parameters for both hydrogen dependent and independent paths of the reaction were determined. The product of the reaction was found to be nitrous oxide.
References
- Belock, C. W.; Cetin, A.; Barone, N. V.; Ziegler, C.J. Inorg. Chem. 2008, 47, 7114 – 7120.
CrossRef - Rees, M. K. Biochemistry 1968, 7(1), 353 – 366.
CrossRef - Rees, M. K. Biochemistry 1968, 7(1) 366 – 372.
CrossRef - Hendrich, M. P.; Petasis, D.; Arciero, D. M.; Hooper, A. B. J. Am. Chem. Soc. 2001, 123(13), 2997- 3005.
CrossRef - Kurnikov, I. V.; Ratner, M. A.; Pacheco, A. A. Biochemistry 2005, 44(6), 1856 – 1863.
CrossRef - Crane, B. R.; Siegel, L. M.; Getzoff, E. D. Biochemistry 1997, 36(40), 12101 – 12119.
CrossRef - Moir, J. W. B.; Wehrfritz, J.; Spiro, S.; Richardson, D. J. Biochem. J. 1996, 319, 823 – 827.
CrossRef - Cabail, Z. M.; Kostera, J.; Pacheco, A. A. Inorg. Chem. 2005, 44, 225 – 231.
CrossRef - Fernandez, M. L.; Estrin, D. A.; Bari, S. E. J. Inorg. Biochem. 2008, 102, 1523 – 1530.
CrossRef - Majee, A.; Kundu,S. K.; Santra, S.; Hajra, A. Tetrahedron Lett. 2012, 53, 4433 – 4435.
CrossRef - Gharah, N.; Chakraborty, S.; Mukherjee, A. K.; Bhattacharyya, R. Inorg. Chem. Acta 2009, 362, 1089 – 1100.
CrossRef - Carneiro, A.; Abreu, A.; Evtuguin, D. V.; Neto,C. P.; Guebitz, G.; Paulo, A. C. J. Mol Cat. B 2000, 9, 293 – 295.
CrossRef - Galli, C.; Gentili, P.; Pontes, A. S. N.; Gamelas, J. A. F.; Evtuguin, D. V. New J. Chem, 2007, 31, 1461- 1467.
CrossRef - Wang, J.; Han, D.; Wang, X.; Qi, B.; Zhao, M. Biosens. Bioelectron. 2012, 36, 18 – 21 .
CrossRef - Mizuno, N.; Misono, M. Chem Rev. 1998, 98, 199 – 218.
CrossRef - Kozhevnikov, I. V. Chem. Rev. 1998, 98, 171 – 198.
CrossRef - Hill, C. L.; Brown, Jr., R. B. J. Am. Chem. Soc. 1986, 108, 536 – 538.
CrossRef - Mansuy, D.; Bartoli, J. F.; Battioni, P.; Lyon, D. K.; Finke, R. G. J. Am. Chem. Soc. 1991, 113, 7222 – 7226.
CrossRef - Dunne, S. J.; Burns, R. C.; Hambley, T. W.; Lawrance, G. A. Aust. J. Chem. 1992, 45, 685 – 693.
CrossRef - Lurie J., Handbookof Analytical Chemistry, Mir Publishers, Moscow, 1975.
- Nipani,S. V.; Gurame, V. M.; Gokavi, G. S. Inorg. Chem. Commun. 2011, 14, 1102- 1106.
CrossRef - Baker, L. C. W.; Weakly, T. R. J. J. Inorg. Nucl. Chem. 1966, 28, 447- 454.
CrossRef - Gurame, V. M.; Supale, A. R.; Gokavi, G. S. Amino Acids, 2010, 38, 789 – 795.
CrossRef - Gurame, V. M.; Gokavi, G. S. Polyhedron, 2008, 27, 1905 – 1910.
CrossRef - Bengtsson, G.; Fronaeus, S.; Bengtsson-Kloo, L. J. Chem. Soc., Dalton Trans. 2002, 2548 – 2552.
CrossRef - Goyal, B.; Prakash, A.; Mehrotra, R. N. Indian J. Chem. 1999, 38A, 541- 546.
- Soni, V.; Mehrotra, R. N. Transition Met. Chem. 2003, 28, 893 – 898.
- Swaroop, R.; Gupta, Y. K. J. Inorg. Nucl. Chem. 1974, 36, 169 – 174.
CrossRef - Saito, A.; Tomari, H.; Choppin, G. R. Inorg. Chim. Acta. 1997, 258, 145 – 153.
CrossRef - Biliski, P.; Motten, A. G.; Biliska, M.; Chignell, C. F. Photochem. Photobiol. 1993, 58, 11- 18.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


