Neutral Gases Adsorption With Hydrogen on Silicon Nanotubes: A Fuel Cell Invistegation
Azin Chitsazan, M. Monajjemi, H. Aghaei and M. Sayadian
Department of Chemistry, Science and research Branch, Islamic Azad University, Tehran, Iran.
Corresponding Author E-mail: Majid_Chiang@yahoo.com
DOI : http://dx.doi.org/10.13005/ojc/330337
In the present study, adsorption of helium, neon, argon and binary mixtures of one of them with hydrogen on (5, 5) armchair silicon nanotube at Temperatures of 50, 100 and 150K and Pressures of 1, 5, 7, 10 and 15MPa were studied. For each binary mixture three different mole fractions of hydrogen were examined. Canonical Monte Carlo simulation by ab-initio calculation was employed for studding the adsorption of above gases on single-walled silicon nanotubes (SW-Si-NTs). The interaction energy of gases with the surfaces of the single Si-NTs obtained from quantum mechanics calculations was fitted to an exact potential functions used for simulating the system. Lennard-Jones potential was used for gas-gas and Morse potential was used for gas-silicon nanotubes interaction. The work was carried out at several temperatures and pressures by using Canonical-Monte-Carlo or CMC simulation in order for studding the effect of temperature and pressure on gas adsorption. The adsorption results showed that by increasing the pressure and decreasing the temperature, the amount of adsorption increases. It was concluded that among the rare gases discussed helium was lowest impact on hydrogen adsorption in a mixture of helium and hydrogen.
KEYWORDS:Molecular simulation; Canonical Monte Carlo Simulation; Morse Potential; Isotherm Adsorption
Download this article as:| Copy the following to cite this article: Chitsazan A, Monajjemi M, Aghaei H, Sayadian M. Neutral Gases Adsorption With Hydrogen on Silicon Nanotubes: A Fuel Cell Invistegation. Orient J Chem 2017;33(3). |
| Copy the following to cite this URL: Chitsazan A, Monajjemi M, Aghaei H, Sayadian M. Neutral Gases Adsorption With Hydrogen on Silicon Nanotubes: A Fuel Cell Invistegation. Orient J Chem 2017;33(3). Available from: http://www.orientjchem.org/?p=33148 |
Introduction
Duo to environmental issues current researches are concentrated on new clean fuels, many studies have been focused on conversion of conventional fossil fuels to hydrogen because of lower pollutant emissions while hydrogen burning.
Considering potentials of the Carbon-Nano-Tubes or SW-CNTs and other non-materials for storage huge fuel gases and basically H21,2, a numerous effort has been conducted, both experimentally3,4 and theoretically5,6 trying for exploring such a possibility3-7.
Carbon nanotubes is an important low-dimensional nanomaterial because of its highly4 interesting properties5, such as narrow diameter6, high aspect ratio8, strong mechanical strength8,9, high chemical and thermal stabilities6-9, excellent heat conduction9-12, interesting electronic and electrical properties and etc7-21 .
Last decade, a few efforts have been focused on the hydrogen adsorption capacities of carbon nanotubes duo to their peculiar electronic and mechanical properties and potentials applications of the hydrogen for fuel12 cell vehicles22-27. However, many of those efforts has been failed for reaching the gravimetric22 densities proposed via the U.S. Department of Energies (6.5 Wt %). Apparently, the designing of a novel material and a modified of porous material significantly increases the adsorption capacities of hydrogen28-30 .
In a contrast of Carbon-based and Silicon-based nanomaterials27, in the terms of their Physico-Chemical properties25, is deeply26 rooted in attributes of the elements and its disparate atomic properties. Generally, Si differs from the C in five points as follows: (1)-lower electronegativity; (2)-kinetically more reactivity; (3) – larger atomic-radius26, and consequently larger orbitals; (4)-smaller energies differences between both S and the P orbitals, and consequently a lower hybridization26 energies; and (5)-the availabilities of energetically30 low-lying “d” orbitals and the abilities for expanding its coordination spheres31 . Those properties of Si and their apparently contrast with C, make Si chemistry and its material so interesting32 and worthy to study33.
One of the main factors that impact the chemicals and physicals properties of Si in comparison with those of carbons are the larger atomic sizes of Si that has longer Si-Si =2.35Å bonds34 in size and weaker bonds with the bond energies of 222,Kj/mol, comparing to carbon-carbon bonds equal 1.54Å including bond energy= 345.6Kj/mol33.
Furthermore, the poor π-π overlapping for Si, Si-Si = 2.16Å with bond energies equal 327Kj/mol are much weaker than the carbon-carbon bonds (1.34Å) with bond energies equal 602Kj/mol34. This disparity leads to a existence35 of varieties in the organic chemistry and nonmaterial like graphite carbon, fullerenes and carbon Nano tubes, whereas36 exist only a few number of stable molecules including Si=Si π bonding2. In the cases of Nano-tubes contrasting with the CNTs, the analogous Si-NTs based on rolled up graphene like sheet seems to be relatively not easily synthesized34, but its existence on a basis of various experimental or theoretical investigation has been reported.
Eberl and Schmidt first have demonstrated the possibilities of the rolling up for a thin Si films30-32 . Moreover to this, Yang has been reported a novel synthesis of the large diameters around ≥50 nm Si-NTs using the chemical vapour deposition methods while using the transmission-electron-microscopy or TEM techniques33 in 2002. After that, Lee35 published a paper related on the growth of pure silicon Nano -tubes using molecular beam epitaxial35 on the porous alumina without any catalysts34. Tang published two works which has been reported the growth of silicon Nano-tubes including a smaller diameter around 13 nm using silicon oxide via hydrothermal synthesis35,36.
To our knowledge, the most interesting results on the synthesis and characterization35 of a thin and presumably single-walled silicon Nano-tubes provided by De Crescenzi, through experimental studies37,38
The successful synthesises of silicon nanotubes caused to a wide investigations for assessing the suitability of silicon Nano-tubes bundles37 as a candidate material for adsorbing efficiently H2. On the aforementioned comparison, between carbon and silicon, silicon has lot electrons in the outer shells, which leads to a higher polarizabilities and stronger dispersion36 forces. Correspondingly, the silicon Nanotubes may show a large and stronger van-der-Waals attraction36 for hydrogen rather than Carbon Nano-tubes. Therefore, it has been reported a multiscale theoretical method for investigating adsorption of instance gases in the silicon Nano-tubes arrays36.
The rest of those papers are organized as follows36-38. (1)-the binding energies between adsorbent and adsorbate obtained from ab-initio calculations were fitted for a propering potential function36. So the result was applied as an input36 in the canonical Monte-Carlo simulation for evaluateing capacities of adsorption of instance gases on SiNT at different pressures, temperatures and mole fractions of mixture. The amount of H2 adsorption in pure and mixture forms on SiNt were also compared to each other.
Methods and Calculations
Computational methods are well placed to a multiscale theoretical approach that combines the first-principle calculation for obtaining the binding energies between instance gases and the Si-NTs and Canonical-Monte-Carlo (CMC) simulation39 for obtaining the weight percentage of pure rare gases and H2 in a mixture of each one of rare gases that might be stored in pure Si-NTs arrays at different thermodynamic conditions.
The first principles quantum chemical calculations applied for investigating of the nature in the gases interactions with the single (5, 5) Si-NTs were accomplished in a DFT system using the package Gaussian0340. Duo to its relatively small diameter, the cylinder cluster model of Si70H20 was selected for SiNT (5, 5) that appears nanotube properties in the lowest possible of length. In the first step, the geometries of the model were optimized. In all of the calculations, the gases molecules and their nearest six Si atoms have been treated with a 6-311++G** basis set, whereas the next atoms were treated with commonly used 4-21G basis set, carried out by the MP1PW9141 exchange-correlation function, which can be provided a more exact dispersion forces rather than B3LYP42,43. On nanotube surface, there are several adsorption sites, depending on the position and orientation of gas molecules: the molecule can be located in the above of Si atoms, the bridge of Si-Si bonds and the centre of silicon hexagon or hollow44. All those cases have been occurred into account for adsorption of all gases in the Si-NTs surfaces. The results have shown among different adsorption position, the hollow44 sites with the H-H bond vertical on the tube surfaces are the most favourable adsorption mode for the outside SiNT wall (see figure 1). This phenomenon has already been observed for adsorption of hydrogen on CNT [30] and SiNT45 surface. The orientation (horizontal or vertical) of gas molecules only considered for hydrogen molecule because the noble gas molecules were described as spherically symmetric.
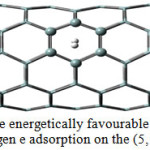 |
Figure 1: The energetically favourable orientations for hydrogen e adsorption on the (5, 5) Si-NTs.
|
An efficient way to implement the results obtains from first-principle calculation in the CMC simulation45, which is an accurate-potential-function to bridge both kinds of calculation.
In this work, the ab-initio potential energies results of the gases with Si-NTs were fitted for a number of usual system ‘functions’, and it has been found that the Morse- potential46 in eq. 1 is the best selection. The parameters in the Morse-potential46 have been obtained according to the fitted binding energies for the molecules in the surfaces which are as close as possible of determining by the first-principle calculations described formerly. It is worth mentioning that the Morse-potential has been used successfully to fit interaction potential computed by the 1th principle calculations in the previous works46-49 .
The interactions between the fluid molecules and the surfaces of the nanotubes were fitted to the Morse- potential, and the form is expressed as follows:
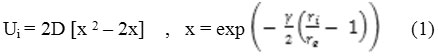
Where ri is the distance between fluid molecule and the tubes surfaces (Å). The values of the constant D, γ and re for gas adsorption outside the nanotube are given in the Table 1.
Table 1: The parameters of Morse-potential used for describing the interactions between the fluid molecules and Si-NTs.
| H2 – SiNT | He – SiNT | Ne – SiNT | Ar – SiNT | |
| _ | _ | _ | _ | |
| D(Kcal/mol) | 0.777 | 0.3373 | 0.8 | 0.9288 |
| g | 5.277 | 6.126 | 5.944 | 6.85 |
| re(Å) | 3.2053 | 3.778 | 3.313 | 3.805 |
As previously mention, we used a CMC-simulation to study the adsorptions of the gases on the (5, 5) SWSiNT. The intermolecular interaction was modelled by the typical 12 – 6 Lennard-Jones pair potential:
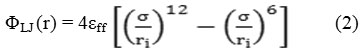
Where, {εffand σff} are the energies and the length parameter in the Lenard Jones potentials, and {r} denoted the centre-towards-centre distances. The subscriptions of {ff} stand for interaction between two section fluid molecules and {ϕLJ} represent the full Lenard Jones potentials.
Cross parameters of the unlike pair i and j particles were obtained using the Lorentz-Berthelot mixing rules:
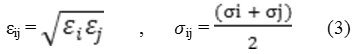
All the interaction potential parameters are given in Table 250-51 . Details of the method can be found elsewhere52,53 .
Table 2: Lennard-Jones potential parameters
|
Interaction |
ε/k5 |
(Å)σ |
|
H2 – H2 |
37 | 2.928 |
| He – He | 10.2 | 2.56 |
| Ne – Ne | 35.6 | 2.75 |
| Ar – Ar | 120 |
3.4 |
The Monte-Carlo simulation is based on the probabilistic phenomenon and concepts. In these methods a system composed of N interacting atoms which are given a set of initial coordinates and orientations. The evolutions of these initial configurations are then generated by successive random displacements of the atoms. The ensemble averages, (f), of the physical quantities f (rN), of function of N particles coordinates, such as the potential energies, are then obtained as an unweighted average over the resulting set of configurations54. Typically between 106 – 105 configurations are generated. In the Monte Carlo approach, no time scales are involved, and the orders in which configurations occur have no particular significance. In most practical applications, the objectives have been estimated Canonical ensemble54 averages, representing the systems composed of fixed number of particles54, N, confined to a fixed volume, V and at a fixed pre-set temperature, T54.
In this work, an armchair type SW-Si-NTs (5, 5) with highly symmetrical structures, with 200 atoms and diameter of 37 Å was selected in our systems. The simulation box used are around 100.00 Å × 100.00 Å × 45.00 Å) contained one SW-Si-NTs. The cut off radius was 2.5 times the collision diameter. The number of the gases molecules in the simulation box (a box with 100.0 Å × 100.00 Å × 45.00 Å dimensions) can be easily calculated at the special temperatures and pressures using virial equation of state54.
Periodic boundaries conditions were applied in all three dimensions54. For each state point, C-M-C simulation consisted of 5×107 configurations that 2×107 steps were to guarantee equilibration and 3×107 steps were to sample the desired thermodynamic properties54.
Furthermore, for obtaining adsorption isotherms, the common parameters were calculated as follows. Gravimetric storage capacities, ρw , were calculated as follows too:
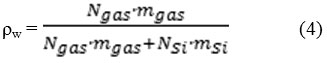
Where NSi and Ngas are the number of Si atoms and gas molecules in our simulation box, and mgas and mSi are the related molar masses, respectively.
Results and Discussion
Adsorption of Pure He, Ne and ar on (5, 5) Sint
The adsorption behaviours of rare gases on (5,5) SiNT, according to eq. (3), are evaluated at one subcritical (50K) and two supercritical (100 and 150K) and pressure ranging between 1 to 15Mpa. The simulation results are exhabited in fig. 1.
All the adsorption isotherms for He, Ne and Ar were characterized by type Ι (Langmuir shape). The intrinsic properties of type Ι isotherms are that adsorptions are limited to a completion of the single monolayer of adsorbate at the adsorbent surfaces. Type Ι isotherms (parabolic behaviour) are visible for the adsorption of gases on microporous55-57 solids whose sizes (pore) are not much larger than the molecular diameters of the adsorbate which complete filling of those narrow pores55. Moreover, adsorption isotherms imply that condensation is prohibited55. Capillary condensation observes when the pores have diameters larger than approximately 2 nm at adequately55 low temperature. It is notable that with a diameter less than 2 nm, no condensations are occurred because the system becomes one-dimensional56, as for SW-CN-Ts. Those explanations are proved with the experimental result for Neon adsorption57 and molecular simulation results for Helium, Xenon and Argon mixture59.
The adsorption isotherms of Helium, Neon and Argon are plotted in figures 1a and 1b as function of pressure at 50K (figure1a) and temperature at 1Mpa (figure 1b). Three He, Ne and Ar adsorption isotherms exhibit similar behaviours. Comparing the amount of He, Ne and Ar adsorbed, it can be seen in figure 1 that the amount of Ar adsorbed is more than that of Ne and the amount of Ne adsorbed is more than that of He at the pressure and temperature range specified55. These are duo to the fact that the gases molecules can varies considerably in the sizes, physical properties, structures and electric properties (dipole and quadruple moments). Hence, Ar has stronger interaction and greater adsorption capacity on SiNTs than Ne that is more than He, under identical conditions.
 |
Figure 1a,b: Asorption isotherms of He, Ne and Ar as function of pressure and temprature, (a) adsorption istherms of He, Ne and Ar in variuos pressure at 50K, (b) adsorption isotherms of He, Ne and Ar in variuos temprature at 1Mpa. Click here to View figure |
Adsorption of H2 in Mixture With Rare Gases on (5, 5) Sint
It is well known that hydrogens are the most promising alternative fuels because of their cleanness and high energies density60,61 . Although it is a gas with critical temperature {33.19K}62, above this temperature hydrogen cannot be liquefied and is unsafe for storage and transported62-84. In this work, we consider hydrogen in binary mixture with each rare gas (He, Ne and Ar) as a solution for safer storage of hydrogen as well as overall the substitution of hydrogen for other flues as fuel cell vehicles.
As we all know, the temperature, pressure and bulk composition play an important role in the adsorption process. For all considered binary mixtures, the gravimetric storage capacities of hydrogens in three mole fraction (xH₂= 0.3, 0.5 and 0.7) are illustrated in figure 2, 3 and 4. To investigate the adsorption behaviour at different pressure and temperature, temperature and pressure were set at 50K (figure 2a, 3a and 4a) and 1Mpa (figure 2b, 3b and 4b) respectively. As we can observe, the variation patterns of H2 adsorption with pressure and temperature is similar to pure He, Ne and Ar adsorption. It is obvious, these calculation indicates that the amount of storage capacity of hydrogen in xH₂= 0.7 is higher than other mole fractions. The corresponding adsorption isotherms are shown in figure 2-4. In addition, it can be shown in the figure 2a that the greatest gravimetric adsorption amount of hydrogen is in xH₂= 0.7 and xHe= 0.3 mixture, duo to the interaction manner of fluid molecules together and fluid molecules with Si-NTs.
 |
Figure 2: adsorption isotherms of Hydrogen/Helium mixture in three mole fractions (xH₂=0.3, 0.5 and 0.7), (a) adsorption isotherms as function of pressure at 50K, (b) adsorption isotherms as function of temperature at 1Mpa. Click here to View figure |
 |
Figure 3: adsorption isotherms of Hydrogen/Neon mixture in three mole fractions (xH₂=0.3, 0.5 and 0.7), (a) adsorption isotherms as function of pressure at 50K, (b) adsorption isotherms as function of temperature at 1Mpa. |
 |
Figure 4: adsorption isotherms of Hydrogen/Argon mixture in three mole fractions (xH₂=0.3, 0.5 and 0.7), (a) adsorption isotherms as function of pressure at 50K, (b) adsorption isotherms as function of temperature at 1Mpa. Click here to View figure |
Comparison of H2/He Mixture and Pure H2 Adsorptions on (5, 5) Sint
Fig. 5 depicts a gravimetric storages capacities for pure hydrogen and 1/3 hydrogen/helium mixtures on the (5, 5) Si-N-Ts as a function of pressure at 50K. As it can be seen from this figure, both isotherms of adsorption are characterized by Langmuir shape, and there is no observation of capillary condensation. Obviously, amount of pure hydrogen adsorption is larger than hydrogen/helium mixture adsorption due to difference between fluid – fluid interaction in mixture and pure condition. In other words, in mixture level of simulation hydrogen molecules not only interact with other hydrogen molecules but have interaction with helium molecule and the number of hydrogen molecules captured by helium molecules. Consequently, hydrogen molecules have lower interaction with SiNT in the mixture level.
In this part, despite the lower amount of stored hydrogen in xH₂= 0.7 and xHe= 0.3 mixture, this system is safer than that of pure hydrogen as a system for storage hydrogen in its applications as fuel cell vehicles.
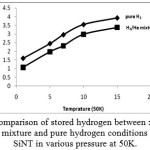 |
Figure 5: comparison of stored hydrogen between xH₂=0.7 and xHe=0.3 mixture and pure hydrogen conditions on (5,5) SiNT in various pressure at 50K.
|
Conclusions
For investigating the effect of pressure, temperature and bulk composition in the adsorption behavior of hydrogens, helium, argon and neon, 5 pressures in the range from 1 – 15 Mpa, three temperatures (50, 100 and 150K) and three mole fraction (0.3, 0.5 and 0.7) in mixture condition were considered. In all studies, the adsorption capacity for adsorption of pure helium, argon, and neon, mixture of each instance rare gases with hydrogen at different mole fractions and pure hydrogen are increasing functions of pressure and decreasing functions of temperatures.
As an amazing result from this research, in xH₂= 0.7 and xHe= 0.3 mixture, despite comparatively larger amount of stored hydrogen in pure hydrogen condition on (5, 5) Si-NTs, this system is safer, i.e. duo to use rare gases, dangerous of explosion in bulk storage of hydrogen decrease in its application as fuel cell vehicles and amount of gravimetric storage capacity in this condition is approximately close the pure hydrogen adsorption.
References
- Iijima S .,Nature, 1991, 354:56.
CrossRef - Lithoxoos GP, Samios J, Carissan YJ , J, Phys Chem C, 2008, 112:16725.
CrossRef - Dillon AC, Jones KM, Bekkedahl TA, Kiang CH, Bethune DS, Heben M J Nature, 1991, 386: 377-379.
CrossRef - Dillon AC, Bekkedahl TA, Jones KM, Heben M J Fullerenes, 1999, 3:716-727.
- Darkrim F, Levesque D J Chem Phys, 1998, 109:4981-4984.
CrossRef - Wang Q, Johnson JK J Phys Chem, B, 1999 103:4809-4813.
CrossRef - Wildoer JWG, Venema LC, Rinzler AG, Smalley RE, Dekker C Nature, 1998, 391:59.
CrossRef - Odom TW, Huang JL, Kim P, Lieber CM Nature, 1998, 391:62.
CrossRef - Charlier JC, Lambin P Phys Rev B,1998 57:R15037.
- Zhou G, Duan WH, Gu BL Chem Phys Lett 2001,333:344.
CrossRef - Hamada N, Sawada S, Oshiyama A Phys Rev Lett , 1992,68:1579.
CrossRef - Saito R, Dresselhaus G, Dresselhaus MS Physical Properties of Carbon Nanotubes. Imperial College Press, London , 1998.
CrossRef - Ouyang M, Huang JL, Cheung CL, Lieber CM Science, 2001 292:702.
CrossRef - Collins PC, Arnold MS, Avouris P Science, 2001 292:706.
CrossRef - Iijima S, Brabec C, Maiti A, Bernholc J J Chem Phys 1996,104:2089.
CrossRef - Thess A, Lee R, Nikolaev P, Dai HJ, Petit P, Robert J, Xu CH, Lee YH, Kim SG, Rinzler AG, Colbert DT, Scuseria GE, Tomanek D, Fischer JE, Smalley RE Science, 1996, 273:483.
CrossRef - Journet C, Maser WK, Bernier P, Loiseau A, DelaChapelle ML, Lefrant S, Deniard P, Lee R, FischerJE Nature, 1997, 388:756.
CrossRef - Mintmire JW, Dunlap BI, White CT Phys Rev Lett 1992,68:631.
CrossRef - Saito R, Fujita M, Dresselhaus G, Dresselhaus MS Appl Phys Lett, 1992, 60:2204.
CrossRef - Saito R, Dresselhaus MG, Dresselhaus MS Phys Rev B, 2000, 61:2981.
CrossRef
- Saito R, Fujita M, Dresselhaus G, Dresselhaus MS Phys Rev B, 1992, 46:1804.
CrossRef - Liu C, Fan YY, Liu M, Cong HT, Cheng HM, Dressethaus MS Science, 1999, 286:1127.
- Zhang XR, Cao DP, Chen JF J Phys Chem B 2003,107:4942.
CrossRef - Wang Q, Johnson JK J Phys Chem B, 1999, 103:4809.
CrossRef - Yin YF, Mays T, McEnaney B Langmuir, 2000 16:10521.
CrossRef - Eric M, Pierre B Langmuir, 2004, 20:7852.
CrossRef - Cao DP, Wang WC Int J Hydrogen Energy, 2007 32: 1939.
CrossRef - Zhao YF, Kim YH, Dillon AC, Heben MJ, Zhang SB Phys Rev Lett, 2005, 95:155504.
CrossRef - Cao DP, Feng PY, Wu JZ Nano Lett, 2004 4:1489.
CrossRef - Mpourmpakis G, Froudakis GE, Lithoxoos GP, Samios J Nano Lett , 2006,6:1581.
CrossRef - Teo BK, Sun XH Chem Re, 2007, 107:1454-1532.
CrossRef - Schmidt OG, Eberl K Nature, 2001 . 410:168.
CrossRef - Sha J, Niu J, Ma X, Xu J, Zhang X, Yang Q, Yang D Adv Mate, 2002, 14:1219-1221.
CrossRef - Jeong SY,Kim JY, Yang HD, Yoon BN, Choi SH, Kang HK, Yang CW, Lee YH Adv Mater,2003, 15:1172-1176.
CrossRef - Chen YW, Tang YH, Pei LZ, Guo C Adv Mater 2005,17:564-567.
CrossRef - Tang YH, Pei LZ, Chen YW, Guo C Phys Rev Lett , 2005,95:116102-116104.
CrossRef - De Crescenzi M, Castrucci P, Scarcelli M, Diociauti M, Chaudhari PS, Balasubramanian C, Bhave TM, Bhoraskar SV Appl Phys Lett, 2005, 86:231901-231903.
CrossRef - Castrucci P, Scarcelli M, De Crecsenzi M, Diociauti M, Chaudhari PS, Balasubramanian C, Bhave TM, Bhoraskar SV Thin Solid Films, 2006 ,508:226-230
CrossRef - Frankel D, Smit B Understanding Molecular Simulation: From Algorithms to Applications. 2nd ed., Academic press, San Diego, 2002.
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T et al Gaussian 03, revision B.0; Gaussian, Inc. Wallingford, CT, 2004.
- Perdew PJ, Wang Y Phys Rev B, 1992 , 45:13244.
CrossRef - Lee C, Yang W, Parr RG Phys Rev B, 1988, 37:785.
CrossRef - Becke AD J Chem Phys, 1993, 98:5648.
CrossRef - Mpourmpakis G, Tylianakis E, Froudakis GE J. Nanosci. Nanotechnol. 2006 , 6:87.
CrossRef - Ryou J, Hong S, Kim G Solid State Communications 2008,148:469-471.
CrossRef - Han SS, Goddard WA J Am Chem Soc, 2007 129:8422.
CrossRef - Smith R, Nock C, Kenny SD, Belbruno JJ, Vece MD, Palomba S, Palmer RE Phys Rev B, 2006, 73:125429.
CrossRef - Jiang AQ, Awasthi N, Kolmogorov AN, Setyawan W, Borjesson A, Bolton K, Harutyunyan AR, Curtarolo S Phys Rev B, 2007, 75:205426.
CrossRef - Vervisch W, Mottet C, Goniakowski J Phys Rev B 2002, 65:245411.
CrossRef - Steele WA Surf Sci, 1973, 36:10651.
CrossRef - Morales JJ, Nuevo MJ J Comput Chem,1995, 16:105.
CrossRef - Allen MP, Tildesley DJ Computer simulation of liquids. Clarendon Press, Oxford, 1987.
- Frenkel D, Smit B Understanding Molecular Simulation: From Algorithms to Applications. Elsevier, New York, 2002.
- Rafii-Tabar H Physics Reports, 2004, 390:235 –452.
CrossRef - Thomas WJ, Crittenden B Adsorption Technology and Design. Elsevier, 1998.
- Cao D, Wu JZ Langmuir, 2004, 20:3759.
CrossRef - Krungleviciute V, Heroux L, Talapatra S, Migone AD Nano Lett, 2004, 4:1133.
CrossRef - Firlej L, Kuchta B Colloids and Surfaces A. Physicochem Eng Aspects, 2004, 241: 149–154.
CrossRef - Greathouse JA, Kinnibrugh TL, Allendorf MD Ind Eng Chem Res, 2009, 48:3425.
CrossRef - Ritter JA, Ebner AD, Wang J, Zidan R Implementing a hydrogen Economy. Mater Today, 2003, 6:18–23.
CrossRef - Elam CC, Padro CEG, Sandrock G, Luzzi A, Lindblad P, Hagen EF Realizing the hydrogen future: the international energy agency’s efforts to advance hydrogen energy technologies. Int J Hydrogen Energy ,2003 28:601–7.
CrossRef - Li Y, Zhao D, Wang Y, Xue R, Shen Z, Li X (Int J Hydrogen Energy, 2006, 32:2513-2517.
CrossRef - Monajjemi, M. Biophysical Chemistry. 2015 207,114 –127.
CrossRef - Bagheri,S.; Moosavi,M.S.; Moradiyeh,N.; Zakeri,M.; Attarikhasraghi,N.; Saghayimarouf,N.; Niyatzadeh,G.; Shekarkhand,M.; Mohammad S. Khalilimofrad, Ahmadin,H.; Ahadi,M.; Molecules 2015, 20, 21636–21657.
- Monajjemi, M. Theor Chem Acc, 2015, 134:77.
CrossRef - Monajjemi, M. Journal of Molecular Modeling , 2014, 20, 2507.
CrossRef - Lee, V.S.; Khaleghian, M.; B. Honarparvar, B.; F. Mollaamin, F. J. Phys.Chem C. 2010, 114, 15315.
- Monajjemi, M. Struct Chem. 2012, 23,551–580.
CrossRef - Monajjemi, M.; Boggs, J.E. J. Phys. Chem. A, 2013, 117, 1670 −1684.
CrossRef - Rahimi, A.;, Oriental Journal of Chemistry, 2016, 32, 6, 2957-296559.
- Monajjemi, M.; Khaleghian, M, Journal of Cluster Science. 2011, 22 (4), 673-692 318.
- Monajjemi, M. Chemical Physics. 2013, 425, 29-45.
CrossRef - Monajjemi, M.; Wayne Jr, Robert. Boggs, J.E. Chemical Physics. 2014, 433, 1-11.
- L Mahdavian, F Mollaamin, B Honarparvar, Fullerenes, Nanotubes and Carbon Nanostructures,2010, 18 (1), 45-55, 2010.
- Raoufi, F.; Aghaee, H.; Oriental Journal of Chemistry, 2016,32, 4, 1839-1858.
- Elsagh, A., Zare, K., Oriental Journal of Chemistry, 2016, 32, 5, 2585-2598.
- Monajjemi, M. Falahati, M.; Mollaamin, F.; Ionics, 2013, 19, 155–164.
CrossRef - Monajjemi, M., Chahkandi, B. Journal of Molecular Structure: THEOCHEM, 2005, 714 (1), 28, 43-60.
CrossRef - Tahan, A.; Mollaamin, F.; Russian Journal of Physical Chemistry A, 2009, 83 (4), 587-597.
- Soofi, N.S., Oriental Journal of Chemistry. 2016, 32, 11, 2327-2345.
- M.; Ketabi, S.; Amiri, A. Russian Journal of Physical Chemistry, 2006, 80 (1), S55-S62.
- Monajjemi, M.; Baheri, H.; Mollaamin, F. Journal of Structural Chemistry.2011, 52(1), 54-59.
CrossRef - Heshmat, M.; Haeri, HH, Biochemistry (Moscow), 2006, 71 (1), S113-S122.
- Khaleghian, M.; Mollaamin, F. Molecular Simulation. 2010, 36, 11, 865– 870.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


