A First Principle Study on the Adsorption of Benzoic Acid onto the (6, 6) and (5, 5) Armchair Single-Walled Carbon Nanotubes
Nosrat Madadi Mahani, Asghar Amiri and Nagmeh Noroozmahani
Department of Chemistry, Payame Noor University, 19395-4697, Tehran, Iran.
Corresponding Author E-mail: nmmadady@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/330309
Benzoic acid (BA) is the typical aromatic monocarboxylic acid often found in domestic and industrial wastewaters. Single-walled carbon nanotubes (SWCNTs), due to their large adsorption capacity, can be used as a new type of adsorbent for the removal of various pollutants from wastewater, such as BA. The (6, 6) and (5, 5) SWCNTs, as adsorbent of the BA compound, have been investigated by theoretical calculations in this study. All calculations have been performed using the DMol3 code, which is based on density functional theory (DFT). The density functional theory was considered within the generalized gradient approximation (GGA) and local density approximation (LAD). The HOMO, the LUMO, energy gap, and electrophilic and nucleophilic attack areas of the reactivity of BA were calculated. The Fukui analysis of descriptor functions shows that the atoms of oxygen are suitable sites for a nucleophilic attack. A molecular dynamics simulation was employed to determine the adsorption behaviour of the BA compound on SWCNTs. The rigid adsorption energy, deformation energy, and adsorption energy were released when the BA was adsorbed on the SWCNTs. The adsorption energy (Eads) values revealed high energy in the BA molecule towards the SWCNT surface. The values of Eads with the two methods were negative, which indicates that the adsorption could occur spontaneously on the surface. The value of Eads was negative, which indicates that the adsorption could occur spontaneously.
KEYWORDS:Benzoic acid (BA); Adsorption energy; Single Walled Carbon Nanotube; Interaction; Density Functional Theory (DFT)
Download this article as:| Copy the following to cite this article: Mahani N. M, Amiri A, Noroozmahani N. A First Principle Study on the Adsorption of Benzoic Acid onto the (6, 6) and (5, 5) Armchair Single-Walled Carbon Nanotubes. Orient J Chem 2017;33(3). |
| Copy the following to cite this URL: Mahani N. M, Amiri A, Noroozmahani N. A First Principle Study on the Adsorption of Benzoic Acid onto the (6, 6) and (5, 5) Armchair Single-Walled Carbon Nanotubes. Orient J Chem 2017;33(3). Available from: http://www.orientjchem.org/?p=34001 |
Introduction
Benzoic acid, the simplest aromatic monocarboxylic acid, is commonly used as a preservative or reaction intermediate besides being employed as an antiseptic agent in food, pharmaceutics, textile and cosmetic industries; therefore, it is often found in domestic and industrial wastewaters1–3. Because of its harmful effects, wastewater must be treated before being sent to water sources. A popular treatment process is the destruction of these compounds by chemical oxidation or biological decaying, and thereafter, elimination through adsorption4. For treatment by adsorption, the primary adsorbents in industrial laboratory usage include activated carbon, alumina, silica, bentonite, peat, chitosan, and ion-exchange resins5. Carbon nanotubes (CNTs) have a very large adsorption capacity and a strong affinity towards all of the kinds of organic pollutants due to their highly spongy and cavity-like structure, light mass density, hydrophobicity, and large specific surface area 6. SWCNTs are forms of carbon with cylindrical nanostructures that can be viewed as a single rolled graphene sheet7. The use of CNTs for the removal of hazardous contaminants from gas phases8–10 and from large volumes of aqueous phases 11–13has been widely investigated with theoretical and experimental methods along with molecular dynamic simulations. The interaction of SWCNT with α-helices has been investigated with molecular dynamic simulations 14. Also, the interaction of DNA bases with carbon nanomaterials has been studied by Hanjun and coworkers 15. The behaviour of adsorption exhibited by folic acid (FA) molecules on SWCNT has been investigated using the atom in molecule (AIM) and natural bond orbital (NBO) analysis by DFT 16. Gao et al. have studied the adsorption of phthalic acid and BA on surface of gold with spectroscopy and DFT calculation methods 17. The interaction between p-aminobenzoic acid and anatase has been investigated using spectroscopy and DFT methods 18. Adsorption of some chlorobenzenes onto the surface of the (5, 5) armchair SWCNT and the graphene sheet has been investigated using DFT calculations and the AIM analysis 19. This work includes a theoretical study of the structural properties and reactivity of the BA molecule and its behaviour when a single molecule is adsorbed on (5, 5) and (6, 6) SWCNT using a DMol3 code and the double numerical basis set with polarization functions (DNP). Based on Fukui indices, the analysis of electrophilic sites were determined. A molecular dynamics simulation was employed to obtain the adsorption behaviour of the BA compound on the (5, 5) and (6, 6) SWCNT.
Methods
All calculations were performed using DMol3 code 20, which is based on DFT. The double numerical plus d-function (DND) on all the electron basis sets was used for all the calculations. The DND basis set involves one numerical function for each occupied atomic orbital, a second set of functions for valance atomic orbitals, and a polarization d-function on all atoms. The density function was treated within the generalized gradient approximation (GGA) with an exchange correlation potential represented by the Wang and Perdew method (GGA-PW91) 21, while local-density approximations (LDA) assumed that the value of the electronic density at each point in space was uniform 22. The gamma-centred k-point was used to represent the Brillouin zone and armchair (6, 6) and (5, 5) SWCNTs with open edges were considered. The diameter of the armchair (6, 6) SWCNT was 8.14 Å and the average bond length was 1.42 Å. Also, the diameter of the armchair (5, 5) SWCNT was 6.78 Å, and the average bond length was 1.42 Å. Five layers of carbon rings along the tube axis (z axis) were modelled, where the bottom-hanging bonds were saturated by hydrogen atoms to simulate the bulk properties. Full geometric optimization was carried out for BA and for armchair (6, 6) and (5, 5) SWCNTs in their original gas phase. The attaching sites of the BA molecule to the SWCNTs by fukui functions were defined. This study focuses on the adsorption of BA to SWCNTs. In the current study, BA molecules were simulated as adsorbate on the armchair SWCNT surface. The molecular dynamic (MD) simulations were performed using the Materials Studio software 23.
Results and Discussions
Using optimized geometries, the energy of the Highest Occupied Molecular Orbital (EHOMO), the energy of the Lowest Unoccupied Molecular Orbital (ELUMO), the total energy, the binding energy, the energy gap, (E), and the total negative charge (TNC) 24, 25 were calculated for BA, (5, 5) and (6, 6) SWCNT molecules (see table 1). The Fukui indices enable the recognition of the reactive regions, the electrophilic and nucleophilic behaviour of a molecule, and its chemical reactivity. For a limited system, when a molecule is accepting electrons, it has the Fukui index for nucleophilic attack (f+), whereas, when the molecule is donating electrons, it has the Fukui index for electrophilic attack (f–). Therefore, the HOMO and the LUMO are related to the negative and the positive Fukui indices, respectively. The Fukui functions permit a forecast of the location of most of the electrophilic and nucleophilic sites of the drug molecules.
Table 1: Quantum chemical descriptors for Benzoic Acid, SWCNT(6.6) and SWCNT(5.5)
| Benzoic Acid | SWCNT(5.5) | SWCNT(6.6) | ||||
| Descriptors | LAD | GGA | LAD | GGA | LAD | GGA |
| Total charge negative | -1.024 | -1.9149 | -5.006 | -2.1399 | -3.9729 | -5.088 |
| HOMO(Hartree) | -0.2635 | -0.2396 | -0.1554 | -0.1497 | -0.1498 | -0.1408 |
| LUMO (Hartree) | -0.1073 | -0.0669 | -0.1214 | -0.1159 | -0.1352 | -0.1293 |
| Total energy((Hartree) | -417.43 | -417.44 | -3789.9 | -3822.7 | -5455 | -5502.2 |
| Binding energy((Hartree) | -3.2295 | -3.2547 | -33.853 | -30.373 | -48.583 | -43.604 |
| Gap Energy (Hartree) | -0.3708 | -0.0909 | -0.2768 | -0.2656 | -0.2821 | -0.3338 |
The HOMO, the LUMO, and the Fukui functions for the studied drugs in the gas phase are shown in Figures 1 and 2, respectively. Figure 1 shows that the LUMO is distributed almost throughout the molecule while the HOMO has distributed across this molecule. In figure 2, the O atoms are the nucleophilic sites of the molecule in the gas phase and the Fukui indices (f–) are spread throughout the entire molecule. This figure shows that the Fukui indices (f+) are spread almost throughout the entire molecule. The HOMO and the LUMO energy distributions depend on the molecular chemical structure of the BA and the SWCNTs. Many electronic properties of chemical structures are directly related to the HOMO and the LUMO levels; thus, the energy gap depends on the energy-geometric structure of the molecule. A high LUMO–HOMO energy gap represents greater stability and low reactivity of the chemical species. The calculations show that this molecule has a high value of the total negative charge. This property characterizes a molecule’s capability of donating electrons. The adsorption of the molecules onto the SWNT surface is enhanced at higher TNC values. Also, the quantum chemical descriptors for SWCNTs are listed in Table 1. The CNTs consist of delocalized electrons. The distribution and character of these electrons depend on the curvature of the CNTs. In the case of (10, 10) CNT, the curvature on the outer surface is less than that of the inner wall. Hence, this situation introduces extra character to the inner wall. In the BA molecule, the active site of binding is essentially determined by unpaired electrons in oxygen atoms. In the non-covalent interaction of BA with SWCNT, the Π- Π stacking interaction plays a major role. In the present case, there was a strong Π- Π interaction between the unpaired electrons in BA and the electrons with extra Π character present in SWCNT. The Monte Carlo simulation process tries to find the lowest energy for the whole system. Using the adsorption locator simulation module distributed by Accelrys 23, the BA molecule–SWCNT configuration was sampled from a canonical ensemble. In the canonical ensemble, the loading of the BA molecule on the SWCCNT substrate was fixed along with the temperature. Monte Carlo simulations help in finding the most stable adsorption sites on SWCNT surfaces by finding the low-energy adsorption sites on both periodic and non-periodic substrates or to investigate the preferential adsorption of the mixtures of adsorbate components26. The adsorption BA molecule on the surface of a (5, 5) SWCNT in figure 3 is shown. Also, the values of adsorption energies by the Monte Carlo simulation are listed in table 2. The total energy is defined as the sum of the energies of the adsorbate components, the rigid adsorption energy, and the deformation energy.
Table 2: calculated energies by the Monte Carlo simulation(kJ/mol)
| SWCNT | Adsorption energy | Rigid Adsorption energy | Deformation energy | |
| (6,6) | GGA | -7.5961 | -0.0357 | -7.5603 |
| LDA | -9.8597 | -0.032 | -9.8276 | |
| (5,5) | GGA | -7.655 | -0.0944 | -7.5606 |
| LDA | -9.8332 | -0.0055 | -9.8277 |
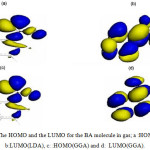 |
Figure 1: The HOMO and the LUMO for the BA molecule in gas; a :HOMO(LDA), b:LUMO(LDA), c: :HOMO(GGA) and d: LUMO(GGA). |
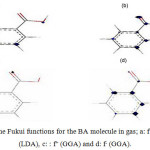 |
Figure 2: The Fukui functions for the BA molecule in gas; a: f+(LDA), b: f– (LDA), c: : f+ (GGA) and d: f– (GGA). |
In addition, adsorption energy is released when the relaxed adsorbate component is adsorbed on the substrate. The adsorption energy is defined as the sum of the rigid adsorption energy and the deformation energy for the adsorbate components. The rigid adsorption energy is released when the unrelaxed adsorbate components are adsorbed on the substrate while the deformation energy is released when the adsorbed adsorbate components are relaxed on the substrate surface. The adsorption energy (Eads) of the BA compound on the surface of (5, 5) and (6, 6) SWCNT were −7.6 and −9.8 kcal/mol with GGA and LDA, respectively. Therefore, adsorption is an efficient treatment process for the removal of BA from industrial wastewater for water treatment.
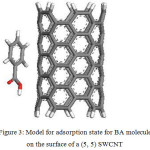 |
Figure 3: Model for adsorption state for BA molecule on the surface of a (5, 5) SWCNT Click here to View figure |
Conclusion
CNTs can be used as suitable absorbents for this process. In this report, we have investigated the structure and the adsorption of BA on SWCNT with the DFT calculation. The interaction between BA and (5, 5) and (6, 6) SWCNT was investigated with the DFT approach using the DMol3 code, a widely used first principles method. The BA molecule is attached both covalently and non-covalently on the walls of CNTs. This work concentrates on the non-covalent adsorption of the BA molecule on SWCNT. The negative adsorption energies demonstrate a more favourable interaction between BA and CNTs. Adsorption is a physisorption process that takes place due to weak Van der Waals interaction between the nanotubes and BA. This process does not require any activation energy and takes place at a low temperature.
Acknowledgements
I gratefully thank Payam Noor University for financial support.
Refrencess
- Chern J. M.; Chien Y. W. Ind. Eng. Chem. Res. 2001, 40, 3775–80.
CrossRef - Howe-Grant M.; Othmer K. Encyclopedia of Chemical Technology. John Wiley & Sons, New York, 1992, 4, 103–115.
- Koczon P.; Dobrowolski J.C.Z.; Lewandowski W.; Mazurek A.P. J. Mol. Struct. 2003, 655, 89–95.
CrossRef - Castilla C. M. Carbon, 2004, 42, 83-94.
CrossRef - McKay G. Use of Adsorbents for the Removal of Pollutants from Wastewaters. CRC Press, New York, 1996, 59–99.
- Baughman R. H.; Zakhidov A.A.; de Heer W. A. Science, 2002, 297,787–92.
CrossRef - Dresselhaus M.; Dresselhaus G.; Eklund P.; Saito R. Phys. World, 1998, 11, 33-38.
CrossRef - Long R.Q.; Yang R.T. Ind. Eng. Chem. Res. 2001, 40, 4288–91.
CrossRef - Long R.Q.; Yang R.T. J. Am. Chem. Soc. 2001, 123, 2058–59.
CrossRef - Agnihotri S.; Rood M.J.; Rostam-Abadi M. Carbon, 2005, 43, 2379–88.
CrossRef - Di Z.C.; Li Y.H.; Luan Z.K.; Liang J. Adsorp. Sci. Technol. 2004, 22, 467–74.
CrossRef - Tan X. L.; Fang M.; Chen C.L.; Yu S.M.; Wang X.K. Carbon, 2008, 46, 1741–50.
CrossRef - Wang S.G.; Gong W.X.; Liu X.W.; Yao Y.W.; Gao B.Y.; Yue Q.Y. Sep. Purif .Technol. 2007, 58, 17–23.\
CrossRef - Balamurugan K. ; Gopalakrishnan R. ; Raman S.S.; Subramanian V. J. Phys. Chem. B, 2010, 11, 14048-58.
CrossRef - Hanjun S.; Jinsong R.; Xiaogang Q. Acc. Chem. Res. 2016, 49, 461–470.
CrossRef - Hamedani S.; Moradi S.; Aghaie H. Chinese J. Struct. Chem. 2015, 34, 1161-69.
- Gao J.; Hu Y.; Li S. ; Zhang Y.; Chen X. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 2013, 104, 41-47.
CrossRef - Thomas A.G.; Jackman M.J.; Wagstaffe M.; Radtke H.; Syres K.; Adel J.; Levy A.; Martsinovich N. Langmuir, 2014 , 21, 12306-14.
CrossRef - Balamurugan K.; Subramanian V. J. Phys. Chem. C, 2013, 117, 21217–27.
CrossRef - Delley B. J. Phys. Chem. 1996, 100: 6107-6115.
CrossRef - Hua X.; Chen X.; Goddard III W.A. Phys. Rev. B, 1997, 55, 16103-108.
CrossRef - Carlos F.; Fernando N.; Miguel M. A primer in density functional theory. Springer, 2003, 60-90.
- Accelrys to Release Enhanced Suite of Chemicals and Materials Modeling and Simulation Tools with Materials Studio(R) 4.1, in: Business Wire, New York, United States, New York 2006. http://accelrys.com/products/collaborative-science/biovia-materials-studio.
- Chattaraj P.K.; Sarkar U.; Roy D.R. Chem. Rev. 2006, 106, 2065-91.
CrossRef - Hazarika K.K.; Baruah N.C.; Deka R.C. Struct. Chem. 2009, 20, 1079-85.
CrossRef - Khaled K.F. J. Solid State Electrochem. 2009, 13, 1743-56.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


