Experimental and Theoretical Study of the Substituted (Η6-Arene)Cr(CO)3 Complexes.
Agus Abhi Purwoko and Saprizal Hadisaputra
Department of Chemistry Education, Faculty of Science and Education, University of Mataram, Indonesia.
Corresponding Author E-mail: agus_ap@unram.ac.id
DOI : http://dx.doi.org/10.13005/ojc/330218
Article Received on : March 16, 2017
Article Accepted on : April 14, 2017
Synthesis of arenetricarbonylchromium(0) complexes, [(η6-arene)Cr(CO)3], has been carried out, wherein arene were benzene (Ph), chlorobenzene (PhCl), phenyltrimethylsilane (PhSiMe3), and acenaphthene (PhNp). Characterization of the compounds was carried out using NMR, IR and UV-visible spectrophotometers. Electronic absorption of these complexeswere measured in various solvents namely methanol, methylene chloride, chloroform, benzene, and isooctane. The complexes showed the electronic absorption of the lowest in the energy range of 313-320 nm, with a relatively high intensity. Density functional theory at the B3LYP/LanL2DZ level of theory was also used to study the geometry parameters, binding energy (BE), vibrational spectra, electronic spectra, frontier molecular orbital (NBO analysis), charge transfer (CT) of the complexes. It was found that the order of the complex stability is: (PhSiMe3)Cr(CO)3> (Ph)Cr(CO)3> (PhNp)Cr(CO)3> (PhCl)Cr(CO)3. The NBO analysis showed that the stability of the complexes arising fromintramolecular interactions and electron delocalization in which synergistic interaction occurs in the arenehyperconjugative orbital ring for metal antibonding orbital and back donation (via metal bonding orbital to bond antibonding orbital ring). The electronic spectrum shows the charge transfer is dominated by ligand to metal charge transfer (LMCT) transition, except for (PhNpe)Cr(CO)3that is dominated by metal to ligand charge transfer (MLCT) and only a small portion is set to d-d transition.
KEYWORDS:Arenetricarbonylchromium; charge transfer transition; DFT; TD-DFT; NBO
Download this article as:| Copy the following to cite this article: Purwoko A. A, Hadisaputra S. Experimental and Theoretical Study of the Substituted (Η6-Arene)Cr(CO)3 Complexes. Orient J Chem 2017;33(2). |
| Copy the following to cite this URL: Purwoko A. A, Hadisaputra S. Experimental and Theoretical Study of the Substituted (Η6-Arene)Cr(CO)3 Complexes. Orient J Chem 2017;33(2). Available from: http://www.orientjchem.org/?p=31668 |
Introduction
Arenechromiumtricarbonyl compounds have been explored extensively and are very versatile agents [1-9]. This compound has many applications ranging from organic synthesis, molecular switches, tunable molecular cables, optical information storage devices and nonlinear optical materials [10-12]. Many earlier studies have explored preparation of arenachromiumtricarbonylcompounds, yet, their syntheses remain an ongoing challenge. The presence of electron withdrawing substituent on the benzene ring deflects π-electron density of the arene, therefore the arene fails to react with the chromium hexacarbonyl;several arenes with electron withdrawing substituentsdo not undergo complexation, e.g. benzoic acid, benzaldehyde, nitrobenzene and benzonitrile[13].
The polysubstitutedcomplex reported in this paper are (benzene)Cr(CO)3,(chlorobenzene)Cr(CO)3, (acenaphthene)Cr(CO)3, and (phenyltrimethylsilane)Cr(CO)3. Earlier study explored the nature of the lowest excited states of these complexes, in which it was reported that the lowest absorption energy band consist with a mixture of ligand field (LF) and metal to arene charge transfer (MArCT) transitions [14]. Unfortunately, the definite order of these transitions could not be resolved well experimentally. This study reported the synthesis and quantum chemical study of polysubstituted (η6-arene)Cr(CO)3 complexes.The quantum chemical study includes geometry molecules, electronic populations and electronic structures of the complexes. The theoretical analysis was performed for optimized molecular geometry, frontier molecular orbital by using the natural bond orbitals scheme, vibrational frequencies, and electronic absorption spectra. The latter was used to identify the nature of the interactions between metal and the ligands in terms of the intensity of metal to ligand or ligand to metal charge transfer (MLCT/LLCT). The electronic absorption spectra were calculated by time dependent density functional theory (TD-DFT) to interpret the UV–Vis spectra obtained at an experimental level.
Experimental
All reagents were commercially available and were used without further purification.
Synthesis
In general, the chromium tricarbonyl complexes were synthesized by refluxing the same amount of molesof chromium hexacarbonyl and appropriate aromatic ligands in aninert solvent for 9 – 48 hours depending on the arene ligands. The mixtures were then filtered using alumina column, followed by rotatory evaporation, and recrystallization. The complexes are obtained in the form of robust solids which have the color ranging from yellow to orange. They are relatively stable in air at room temperature.
Physical Measurements
The infrared spectra were recorded on a Perkin-Elmer 283B spectrophotometer using KBr pellet. Electronic spectra were measured on UV-Vis (HP 8450A) spectrophotometer in methanol, benzene, chloroform, dichloromethane, and isooctanesolution. 1H NMR spectra were obtained at room temperature in CDCl3 using a Bruker AM 360spectrometer.
Computational Method
All calculations have been performed with the GAUSSIAN-03 program [15]. Geometry optimizations utilized the B3LYP method [16-18] and the LANL2DZ basis set [19]. Previous studies have shown this to be an appropriate level of theory for chromium tricarbonyl complex of arenes[20-21]. NBO [22-24] calculations have been done at B3LYP/LANL2DZ level.
Results and Discussion
The structures of arenechromiumtricarbonylcomplexes were optimized at the density functional level of theory using the B3LYP/LanL2DZ methodology. The different typesof substituents were added into the benzeneunit as depicted in Scheme 1. Figure 1 depicts the mode of substitution of the benzene unit, which was performed with all of the aforementioned moieties.
The geometrical properties of the complexes show that the structural changes in (η6-arene)Cr(CO)3 complex induced by introducing different substituent groups is very small (Fig. 1) owing to the tripodC-O bonds making the molecule rigid. This leads to a small number of conformational isomers, and as a result the computational effort is also reduced.The calculated geometry is in a good agreement with the X-ray structural measurements of (Ph)Cr(CO)3. Thecalculated Cr-O bond distances for all complexes are relatively similar range from 1.8293 Å to 1.8386 Å. In the complex of (PhNp)Cr(CO)3 the position of Cr(CO)3 tripod 0.17 Å shifts from the ring centertoward the most external C-C bond (parallel to the bond between the two rings). The distance of chromium to the ring centerfor (PhNp)Cr(CO)3 is about 1.80 Å, a slightly longer distance compared to benzene-Cr interaction at (Ph)Cr(CO)3 of 1.720 Å (exp.: 1.724-1.726 Å)[25].
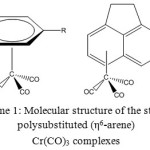 |
Scheme 1: Molecular structure of the studied polysubstituted(η6-arene) Cr(CO)3 complexes |
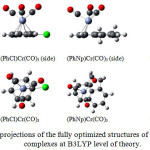 |
Figure 1: Two projections of the fully optimized structures of (η6-arene)-Cr(CO)3 complexes at B3LYP level of theory.
|
Table 1: Geometric parameters of the fully optimized structures of (η6-arene)Cr(CO)3 complexes at B3LYP level of theory.
| Geometrical Parameters | Complexes | |||
| Bond Length (A) | (Ph)Cr(CO)3 | (PhCl)Cr(CO)3 | (PhNp)Cr(CO)3 | (PhSiMe3)Cr(CO)3 |
| Cr-O12(10) | 1.8333 | 1.8386 | 1.8265 | 1.8281 |
| Cr-O13(11) | 1.8354 | 1.8354 | 1.8314 | 1.8293 |
| Cr-O14(12) | 1.8386 | 1.8339 | 1.8203 | 1.8304 |
| Cr-C1 | 2.3183 | 2.3183 | 2.3530 | 2.3015 |
| Cr-C2 | 2.3172 | 2.3172 | 2.4035 | 2.3469 |
| Cr-C3 | 2.3281 | 2.3281 | 2.4586 | 2.3027 |
| Cr-C4 | 2.2985 | 2.2985 | 2.3266 | 2.3260 |
| Cr-C5 | 2.3124 | 2.3124 | 2.2842 | 2.3188 |
| Cr-C6 | 2.2980 | 2.2980 | 2.3269 | 2.3243 |
Hexacarbonylchromium(0), Cr(CO)6, is a reactant in the synthesis of (arene)Cr(CO)3complexes. In the Cr(CO)6 molecule, Cr-C-O bond has a linear shape and structure of the Cr-C-O can easily be identified on the IR spectrum that showed CO stretching mode in the 1850-2125 cm-1[26]. The unique identity of (arene)Cr(CO)3 is the presence of two high-intensity IR absorption which are stretching modes of COligand; the energy absorptionsare in the area 2000 – 1960 cm-1, and in the area of energy from 1910 to 1870 cm-1. Assuming C3vsymmetry locally on the cluster of Cr(CO)3, the two uptake is thought to originate from vibrations of symmetric nondegenerate (A1) and antisymmetric double degenerate (E) [27]. IR spectra and 1HNMR spectra for all of four chromium tricarbonyl complexesare presented in Table 1.
X-ray crystallography and neutron diffraction [28] concluded that the C – C in the arene ring elongated compared to its native form (prior coordinated with the group Cr(CO)3. This phenomenon is interpreted as a form of a reduction in the degree of aromaticity of the ring of arene after coordination with chromium group oftricarbonyl[29]; an explanation that is consistent with the results of1HNMR spectra in this study. Interaction ofCr(CO)3 with the arene ring reduceselectron density in the ring [30]. This results in lower degrees of aromaticity, manifested in the 1HNMR spectra that indicate the presence of hydrogen aromatic shift into a higher energy position (up field) (δ = 4.6 to 5.6 ppm, while the free aromatic ranging from 6.0 to 7.0 ppm).
Table 2: Experimented and calculated 1H NMR and IR spectra of the (η6-arene)Cr(CO)3complexes
| Complexes | 1H-NMR, δ (ppm)a | IR, νCO, cm-1 (lit)b | ||||
| H aromatic | H substituent | A1 | E | |||
| (Ph)Cr(CO)3 | 5,4 (s) | c | 1965 (1974)d(1939)f | 1887 (1898) (1878)f | ||
| (PhCl)Cr(CO)3 | 5,4 – 5,5 (m) | c | 1980 (1992)e(1947)f | 1922 (1931)e(1981)f | ||
| (PhNp)Cr(CO)3 | 7,3 – 7,7 (m) | 3,4 (s) | 1968 (1928)f | 1871 (1868)f | ||
| (PhSiMe)Cr(CO)3 | 5,2 – 5,6 (m) | 0,3 (s) | 1980 (1934)f | 1904 (1875) | ||
a) The spectra were taken in CDCl3 solution; using TMS as an internal standard; (S) = singlet, (m) = multiplet
b) Stretching frequencies of CO relative to polystyrene. Using solvents CDCl3
d) In CHCl3[30]
e) In cyclohexane [30]
f) Calculated IR spectra
A characteristic feature of electronic absorptions of arenetricarbonylchromium(0) complexes is the maximum absorbtion in the region 313-320 nm with high intensity (ε ~ 0.97 x 104 up to 2.04 x 104 M-1cm-1), except for (PhNp)Cr(CO)3complex, that has λmax at 352 nm with an intensity that is almost the same (ε ~ 0.91 x 104 M-1cm-1).This indicates a transition from metal to ligand and ligand to metal charge transfers (MLCT/LMCT).
Table 3: Electronic absorbtion data of (η6-arene)Cr(CO)3complexes in a variety of solvent-saturated N2 at 298 K.
|
Complexes |
Absorbance maximum (λmax, nm) | |||||||||
| CH3OH | CH2Cl2 | CHCl3 | C6H6 |
C8H18 |
||||||
| (Ph)Cr(CO)3 | 312 | 313 | 315 | 313 | 313 | |||||
| (PhCl)Cr(CO)3 | 320 | 319 | 320 | 320 | 318 | |||||
| (PhNp)Cr(CO)3 | 344 | 347 | 350 | 352 | 353 | |||||
| (PhSiMe3)Cr(CO)3 | 315 | 316 | 318 | 316 | 316 | |||||
In order to assign the already reported experimental electronic absorbtion bands of (Ph)Cr(CO)3, (PhCl)Cr(CO)3, (PhNp)Cr(CO)3, and (PhSiMe)Cr(CO)3, the TD-DFT calculations have been carried out on these complexes in methanol at the B3LYP/GEN level of theory. TD-DFT is a useful method for studying excitation energies, and its application has increased in the recent years [31]. This method does give some errors in the excitation energies of charge-transfer states, however; better results may be obtained by using hybrid functionals which include a mixture of exact Hartree-Fock exchange with DFT exchange correlation [32]. The calculated electronic excitation energies and transition wavelengths (λcal) of the complexes, along with their oscillator strengths, assignments and transitions with significant coefficients of the wavefunction are listed in Table 4, along with the experimental transition wavelengths (λexp).
Table 4: Caculation of electronic absorbtions of (Ph)Cr(CO)3, (PhCl)Cr(CO)3, (PhNp)Cr(CO)3, and (PhSiMe3)Cr(CO)3complexes, in Methanol, from TD-DFT calculations at the B3LYP/LanL2DZ.
| Complexes | Electronic transition | Transition Coefficient(c) | Weight % of Transition | Oscillator Strength | Excitation Energy | λcal | scaled λcal | λexp | Assignment |
| (Ph)Cr(CO)3 | H →L | 0.471 | 22.2 | 0.0001 | 3.288 | 377 | 271 | MCT/ILCT | |
| H → L+3 | 0.387 | 15.01 | 0.0007 | 3.363 | 368 | 265 | d-d/MLCT/LMCT/LLCT/ILCT | ||
| H → L+3 | 0.185 | 3.42 | 0.0051 | 3.446 | 359 | 259 | 312 | LMCT/ILCT | |
| (PhCl)Cr(CO)3 | H → L | -0.23 | 5.09 | 0.0002 | 3.227 | 384 | 276 | LMCT/ILCT | |
| H-2 →L+2 | 0.384 | 14.75 | 0.0043 | 3.290 | 360 | 259 | 320 | LMCT/ILCT | |
| H-1→ L | -0.240 | 5.53 | 0.0000 | d-d/MLCT/LMCT/LLCT/ILCT | |||||
| H-1→ L | 0.374 | 13.97 | 0.0009 | 3.688 | 336 | 242 | d-d/MLCT/LMCT/LLCT/ILCT | ||
| (PhNp)Cr(CO)3 | H→ L | 0.617 | 38.02 | 0.0056 | 2.736 | 453 | 326 | LMCT/ILCT | |
| H-2→ L | 0.559 | 31.27 | 0.0240 | 2.906 | 426 | 307 | 344 | MLCT /LMCT/ILCT | |
| H → L+2 | 0.474 | 22.50 | 0.0074 | 3.390 | 365 | 263 | LMCT/ILCT | ||
| (PhSiMe3)Cr(CO)3 | H-1 → L+2 | 0.421 | 17.74 | 0.0001 | 3.297 | 376 | 270 | LMCT/ILCT | |
| H-1 → L+1 | 0.591 | 34.90 | 0.0030 | 3.632 | 341 | 245 | 315 | LMCT/ILCT | |
| H-2 → L+2 | 0.273 | 7.44 | 0.0000 | d-d/MLCT/LMCT/LLCT/ILCT | |||||
| H → L | 0.288 | 8.29 | 0.0002 | 3.809 | 325 | 234 | LMCT/ILCT |
Transition metal complexes generally show three types of electronic excitation bands that cover a wide wavelength range: d-d (crystal-field) transitions (300 – 1500 nm); metal-to-ligand charge-transfer (MLCT) and ligand-to-metal charge transfer (LMCT) transitions (200 – 500 nm); transitions that are localized on the ligands commonly known as intra-ligand charge transfer (ILCT) transitions, that regularly occur in the ultraviolet region [33]. ILCT results from n → π* and n → π* transitions and are affected by the type of coordination.
The scaling factor 0.72 was used to correct the calculated singlet-singlet electronic excitation wavelengths [34]. The calculate electronic spectrum of(Ph)Cr(CO)3, (PhCl)Cr(CO)3, and (PhSiMe3)Cr(CO)3 have closely similar bands, between 325-360 nm,that can be attributed mainly to ligandtometal charge transfer transitions and smaller band with low oscillator strength (f ≤ 0.0005), that is due to a d-d transition. The latter assignment was made since d-d transitions usually have very low intensities and lack of solvent dependent. For (PhSiMe3)Cr(CO)3 complex, the band at 426 nm with oscillator strength of 0.0240, is the most intense and attributed to electronic excitation from HOMO-2 to LUMO. This band, therefore, arises from a mixed of MLCT and LMCT charge transfer character.
In general, the binding energy is related to the stability of the complexes. The binding energies for the chromium-arene complexes were calculated as the energy difference between the complexes and their monomer. The calculated binding energies for the complexes are listed in Table 5.
 |
Table 5: Calculated binding energy (BE), Selected second order binding energy (E2), frontier molecular orbital (HOMO-LUMO), and charge transfer (CT) of (η6-arene)Cr(CO)3complexes. Click here to View table |
*Experimental value [35].
The calculated binding energy of the complex are 48.73 kcal.mol-1, 45.64 kcal.mol-1, 45.62 kcal.mol-1, and 41.46 kcal.mol-1 for (PhSiMe3)Cr(CO)3, (Ph)Cr(CO)3, (PhNp)Cr(CO)3, (PhCl)Cr(CO)3, respectively.The (Ph)Cr(CO)3 binding energy is 45.64 kcal.mol-1, not too far from the experimental value, i.e. the benzene-Cr bond in the (Ph)Cr(CO)3 complex for which the binding energy is 43 kcal.mol-1as reported by Brown et al. [35]. The binding energy trend shows that electron withdrawing substituent on the ring reduces the binding energies from the original (Ph)Cr(CO)3. In contrast, the presence of additional two benzene ring on (PhNp)Cr(CO)3act as electron donating groups, and therefore the binding energy increase compared to (Ph)Cr(CO)3binding energy. The binding energy trend is consistent with calculated Mulliken charge transfer trend from ring → Cr.
The stability trend of the complexes is further studied by the second-order binding energies (E2) trend based on the natural bond orbital (NBO) analysis. E2 corresponds to the intensity of interaction between Lewis donor and non-Lewis acceptor NBO[30-32, 36,37]. Table 5 shows a positive correlation of the binding energy with the second-order binding energy. All the complexes have either the bonding (ring → σ*Cr–CO) (or) back bonding (Cr–CO → ring) interactions.The maximum E2 contribution of ring → σ*Cr–COfor(PhSiMe3)Cr(CO)3, (Ph)Cr(CO)3, (PhCl)Cr(CO)3range from 199.28-202.33 kcal with Cr–CO → ringback bonding15.64-16.72 kcal. The difference was found for ring → σ*Cr–COE2 maximum of (PhNp)Cr(CO)3complex (151.35 kcal), where two additional benzene on ligand (acenapthene) may distortinteraction between Lewis donor and non-Lewis acceptor NBO.
Conclusion
The aim of this study was to synthesize tricarbonylchromium(0) arene complexes and to analyse synthesis products using experimental and theory studies. Synthesis products of tricarbonylchromium (0) arene complexes were (Ph)Cr(CO)3, (PhCl)Cr(CO)3, (PhSiMe3)Cr(CO)3, and (PhNp)Cr(CO)3. Characterization by1H-NMR, IR, and UV-visible showthat the complexes were successfully synthesized. DFT study at B3LYP/LanL2DZ level of theory was also used to study the geometry parameters, binding energy (BE), the frontier molecular orbital (NBO analysis), the charge transfer (CT), electronic and vibration spectrum. The electronic spectrum shows the transfer fee is dominated by metal to ligand charge transfer (LMCT) transition, except (PhNp)Cr(CO)3 is dominated by metal to ligand charge transfer (MLCT) and only a small portion is set to d-d transition. It was found that the complex stability order were (PhSiMe3)Cr(CO)3> (Ph)Cr(CO)3> (PhNp)Cr(CO)3> (PhCl)Cr(CO)3. NBO analysis showed that the stability of the complexes arising from hyperconjugative orbital rings for metal antibonding orbital donation and back bonding orbital.
References
- Jaouen, G.. Transition Metal Organometallics in Organic Synthesis: Organic Chemistry: A Series of Monographs, 2016.,33.2, 2, 65.
- Fukin, G. K.; Cherkasov, A. V.; Artemov, A. N. ChemistrySelect,2016,1(15), 5014-5018.
- Wilson .Konderka, C.; Doxtator, K.; Metallinos, C. Adv. Syn. Catalysis, 2016,358(16), 2599-2603.
- Glans, L.; Taylor, D.; de Kock, C.; Smith, P. J.; Haukka, M.; Moss, J. R.; Nordlander, E. J. Inorg.Biochem. 2011. 105(7), 985-990.
- Murai, M.; Uenishi, J. I.; Uemura, M.Org.Lett. 2010,12(21), 4788-4791.
- Gloriozov, I. P.; Marchal, R.; Saillard, J. Y.; & Oprunenko, Y. F. Eur. J.Inorg. Chem.2015, (2), 250-257.
- Murai, M.; Sota, Y.; Onohara, Y.; Uenishi, J. I.;Uemura, M.J. Org. Chem.2013, 78(21), 10986-10995.
- Compain, J. D.; Stanbury, M.; Trejo, M.; Chardon‐Noblat, S. Eur. J.Inorg. Chem.2015, (35), 5757-5766.
- Semenycheva, L.; Artemov, A.; Valetova, N.; Matkivskaja, J.; Moykin, A. J.Inorgan.Organomet.Polym.2016. 26(5), 921-931.
- Arrais, A.; Diana, E.; Marabello, D.; Gervasio, G.; Stanghellini, P. L. J.Organomet. Chem. 2011,696(11), 2299-2305.
- Konietzny, S.; Finze, M.; Reiß, G. J. J. Organomet. Chem, 2010, 695(18), 2089-2092.
- Momoi, Y.; Okano, K.; Tokuyama, H. Synlett, 2014, 25(17), 2503-2507.
- Zhang, J.; Stanciu, C.; Wang,B.; Hussain, M. M.; Da, C. S.; Carroll, P. J.; Walsh, P. J.; J Am Chem Soc, 2011, 133(50), 20552-20560.
- Purwoko, A. A; Lees. A. J.; Indo. J. Chem, 2009, 9(1), 123-126; Purwoko, A. A., Indo. J. Chem, 2008, 8(1), 13-17.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Kudin, K.N.; Strain, M.C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G.A.; Ayala, P.Y.; Cui, Q.; Morokuma, K.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Cioslowski, J.; Ortiz, J.V.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.R.; Gomperts, R.; Martin, L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.P.; Gill, M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Andres, J.L.; Gonzalez, M. Head-Gordon, M.; Replogle, E.S.;. Pople, J.A. GAUSSIAN 03, Revision A.6. 2002, Gaussian Inc., Pittsburgh PA.
- Becke, A. D., J. Chem. Phys. 1993, 98, 1372.
- Becke, A. D., J Chem Phys, 1993, 98:5648
- C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 1988, 37, 785.
- Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82, 299
- Kalpana ,A.; Akilandeswari, L.Comput. Theor.Chem.2016, 1084, 103-108
- Kalpana, A.; Akilandeswari, L. Comput.Theor. Chem.2015. 1069, 125-131.
- Foster, J. P.; Weinhold, F., J. Am. Chem. Soc.,1980, 102 (24), 7211-7218
- Reed, a. D.; Curtiss, L. A.; Weinhold, F.; Chem. Rev. 1988, 88.
- Glendening, E. D.; Reed, A. E.; Carpenter, J. E.; Weinhold, F., 1988, NBO 31 ProgramManual.
- Bailey, M. F.; Dahl, L. F, Inorg Chem,1965, 4(9), 1314-1319.
- Cotton, F. A.; Wilkinson, G, 1988, 5thEd., John Wiley & Sons: New York.
- Davis, R.; Kane-Maguire, L. A. V. 1982, v.3, Pergamon Press: Oxford.
- Rees, B.; Coppens, P.; ActaCrystallographica Section B: Structural Crystallography and Crystal Chemistry, 1973, 29(11), 2516-2528.
- Connor, J. A.;Martinho-Simoes, J. A.; Skinner, H. A.; Zafarani-Moattar, M. T, J. Organomet. Chem.1979, 179(3), 331-356.
- Brown, D. A., Raju, J. R., J. Chem. Soc. A: Inorg. Phys.Theor.1966, 1617-1620.
- Jacquemin, D., Duchemin, I., &Blase, X.J.Chem. Theor. Comput.2015,11(11), 5340-5359.
- Zárate, X.; Schott, E; Carey, D. M.; Bustos, C.;Arratia-Pérez, R.; J. Mol. Structure: THEOCHEM. 2010,957, 126-132.
CrossRef - Zhao, F. Wang, J. X., Wang, Y. B., Comput. Theor.Chem, 2001, 973, 40-46.
- Mohammed, L. S., Hamza, I. S., AL-Deen, F. R. M.; Muhyedeen, B. R. J, J. Applicable Chem, 2014, 3, 2102-2122.
- Brown, D. L. S.; Connor,J. A.; Demain, C. P.; Leung, M. L.; Martinho-Simoes,J. A.; Skinner, H. A., Zafarani-Moattar, M. T., J.Organomet. Chem. 1977, 142, 321-335.
- Hadisaputra, S.;Pranowo, H. D.; Armunanto, R. Indo. J. Chem.2012, 12(3), 207-216.
- Hadisaputra, S.;Canaval, L. R.;Pranowo, H. D.; Armunanto, R. Monats. Chem.2014, 145(5), 737-745.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


