Comparative Study of Decomposition Adsorption of Sarin on Znn on and Cdn on (n=1,4), by Theoretical Method
Farrokh Roya Nikmaram
Department of Chemistry, Faculty of Science, Yadegar - e- Imam Khomeini (RAH) Shahre Rey Branch, Islamic Azad University, Tehran, Iran.
Corresponding Author E-mail: Nikmaram87@yahoo.com
DOI : http://dx.doi.org/10.13005/ojc/330226
Article Received on : November 10, 2016
Article Accepted on : January 11, 2017
The calculations of the electronic and structural properties for interactions of Sarin with ZnnOn and CdnOn (n=1,4) have been conducted by B3LYP/6-31++G(d,p) level of DFT method. The ZnO affects on disconnection of propyl bond of Sarin and Cd4O4 interacts with Fluorine atom of Sarin. The results of Radial Distribution Function (RDF) show that the interaction of Sarin is probable at 380 K with ZnO as covalent bond and at 308 K with Cd4O4 by Van der waals forces.
KEYWORDS:Sarin; ZnnOn; CdnOn; Decomposition; DFT; RDF
Download this article as:| Copy the following to cite this article: Nikmaram F. R. Comparative Study of Decomposition Adsorption of Sarin on Znn on and Cdn on (n=1,4), by Theoretical Method. Orient J Chem 2017;33(2). |
| Copy the following to cite this URL: Nikmaram F. R. Comparative Study of Decomposition Adsorption of Sarin on Znn on and Cdn on (n=1,4), by Theoretical Method. Orient J Chem 2017;33(2). Available from: http://www.orientjchem.org/?p=32104 |
Introduction
Chemical warfare agents (CWAs) have been used in the World Wars that to cause killing and injuring a large number of people [1]. The stable complexes of CWAs with acetylcholine esterase enzyme at cholinergic synapses of nervous systems (in humans) leads to a variety of effects such as hypotension, muscle tremors and convulsions [2]. Sarin is the nerve agent of CWA. It is a derivative of methyl phosphono fluoridate, Fig1. CWAs are far too dangerous for experimental study. Hence, researchers prefer to use theoretical methods to investigate their decomposition possibility. The nano metal oxides with low coordination number [3] have unusual electronic properties and adsorption behavior due to their defect sites (Frenkel & Schottky), Lewis acid (metal cations) sites, Lewis base (oxide anions) sites and, high surface area [4,5]. Decomposition adsorption of Chemical warfare agents (CWAs) on nano metal oxides yields non-toxic products. Dimethyl methyl phosphonate, [DMMP, CH3PO (OCH3)2] is a nontoxic organophosphorus compound and it used as a simulant for CWAs chemical warfare agent. Both experimental and theoretical studies have established that decomposition of DMMP is facilitated on the small cluster of nano metal oxides Al2O3 [6,7], MgO [8], SiO2[9], TiO2[10] , ZnO [11] and Mg4O4 [12] due to their electronic and surface properties. Metal oxides are used as adsorbents, catalysts and catalyst supports and they are environment friendly for decontamination applications, such as decontamination on the battlefield, filtration systems, and decomposition of CWAs [13]. One type of nano metal structure as form of M4O4, is similar to nanocone.
The nanocones are observed as caps on the ends of nanotubes, and also as free standing nanostructures [15]. More recently, a theoretical study by Alfieri and Kimoto has indicated that nanocones with disclination angles 60º ,120º, 240ºand 300ºare stable[16]. The cone is entirely characterized by its cone angle [17].
Jin Chang and Eric R. Waclawik at 2012[18] and A. Bagheri Ghomi at 2016[19] have reported the synthesis of Zn4O4 nanocone. In the current paper, we investigate the M4O4 type of nano metals as a potential candidate for adsorption of Sarin.
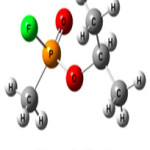 |
Figure 1: Sarin Click here to View figure |
Model and Simulation Details
Studies by Harrison have shown that DFT method with Lee-yang-Parr’s correlation Functional (B3LYP) provides better agreements with experimentally derived band gaps for a wide class of zinc-blend and wurtzite-structured III–V materials [20]. Therefore, in this work, the B3LYP level of DFT has been used for all of the calculations and B3LYP values have been scaled by a factor of 0.96 [21]. But one concern lies in choice of basis sets on the accuracy of the final results. The experimentally rotational constants are related to molecular geometry [22, 23]. Therefore, the rotational constants of the optimized Sarin at B3LYP/6-31G (d), B3LYP/6-31+G(d) and B3LYP/6-31++G(d,p) have been calculated and compared with the experimental values for choice the suitable basis set. The error is being defined as ((Cal-Exp)/Exp) and summed over all three rotational constants.
The calculations of the electronic and structural properties for interactions between the Sarin and ZnO (Figs.2,3), Zn4O4 nanocone (Figs.4,5), CdO (Figs.6,7), and Cd4O4 nanocone (Figs.8,9) have been conducted by DFT method, in conjunction LANL2DZ for metal, as implemented in the Gaussian 09 program package [24].
A small cluster of nanocone containing four Zn atoms (or Cd atoms) and four O atoms with disclination angle equal to 240º and height equal to 4 have been created by Nanotube Modeler 2014 software [25].
In this article, the geometry of the Sarin and MO have been fully optimized while the M4O4 (M=Zn or Cd) has been fully frozen.
We have considered two position of Sarin connections on MO and M4O4 (M=Zn or Cd).
First position (P1) is the connection of oxygen from phosphonyl group (P=O) of Sarin to M of metal oxide.
Second position (P2) is the connection of Fluorine from Sarin to M of metal oxide.
The geometry of connected systems of P1 and P2 complexes has been fully optimized.
For evaluation of basis sets difference for adsorbed systems, the interaction energies (ΔEint) of studied adsorption systems have been corrected by the Basis Set Superposition Error (BSSE), Eq.1.
ΔE int =E (MnOn+ Sarin) – E(MnOn) – E(Sarin) + ΔE(BSSE) Eq.1
that the first term in ΔEint is the energy of the adsorbed system and the next two terms are the energies of the bare MnOn (M= Zn, Cd and n= 1,4) and the free Sarin molecule, respectively, and ΔE(BSSE) is correction of BSSE.
The harmonic vibrational frequencies of the fully optimized structures have been calculated to confirm the stationary point as a local minima with all positive frequencies. The electronic properties of nanostructures have been described by lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) [26].
Results and Discussion
In this work, we have focused on the rotational constants of Sarin for choosing the suitable basis set. The Sarin structure has been optimized at B3LYP level of DFT with 6-31G(d), 6-31G+(d) and 6-31++G(d,p) basis sets. The results for the rotational constants in Table 1 shows that the average error of calculated rotational constants are about 8.4% for B3LYP/6-31G(d), 11% for B3LYP/6-31G+(d) and 3.5% for B3LYP/6-31++G(d,p). Therefore, the calculated rotational constants by B3LYP/6-31++G(d,p) with the smallest error are in more agreement with the experimental values with respect to other. Thus, we are focused on the B3LYP/6-31++G (d,p) results for the next calculations.
The structural stability of nanostructures can be described by calculated energy. In this work, the interaction between Sarin and MnOn (M=Zn or Cd and n=1, 4) has been studied for two position of connection. The P1 Position indicates the interaction of oxygen from P=O group of Sarin MnOn. The P2 position is related to interaction of Fluorine of Sarin with metal from MnOn. Table 2 presents the corrected interaction energies, dipole moments, bond lengths and natural charge of oxygen and metal for all the interacting systems.
Table 1: Calculated and Experimental Rotational Constants (MHz) for Sarin
|
Methods |
A |
B |
C |
Sum of |
|
errors |
||||
|
Experimental23 |
2874.07 |
1168.57 |
1056.33 |
0 |
|
B3LYP/6-31G(d) |
2770.95 |
1136.1 |
1032.19 |
0.084 |
|
B3LYP/6-31+G(d) |
2764.28 |
1120.73 |
1022.18 |
0.11 |
|
B3LYP/6-31++G(d,p) |
2766.62 |
1125.05 |
1021.27 |
0.035 |
The interaction energy of Sarin with ZnO,P1 (Fig.2), ZnO,P2 (Fig.3), Zn4O4 ,P1 (Fig.4), Zn4O4 ,P2(Fig.5), CdO,P1( Fig.6), CdO,P2( Fig.7), Cd4O4 ,P1 (Fig.8) and Cd4O4 ,P2 (Fig.9) have been determined to be – 19.73, -9.31, -35.06, -21.23, -15.03, -8.81, -37.11and -24.50 Kcal/mol, respectively. The values of ΔEint show that the connection between the phosphonyl O atom of Sarin with MnOn (n=1 or 4) (all of P1 positions) is energetically favored over Fluorine connecting (P2 positions).
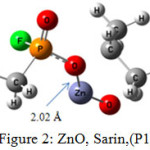 |
Figure 2: ZnO, Sarin,(P1) Click here to View figure |
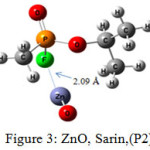 |
Figure 3: ZnO, Sarin,(P2) Click here to View figure |
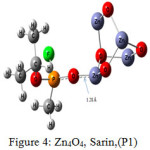 |
Figure 4: Zn4O4, Sarin,(P1) Click here to View figure |
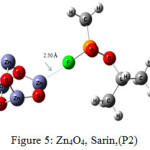 |
Figure 5: Zn4O4, Sarin,(P2) Click here to View figure |
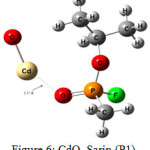 |
Figure 6: CdO, Sarin,(P1) Click here to View figure |
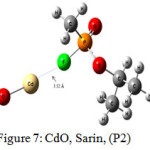 |
Figure 7: CdO, Sarin, (P2) Click here to View figure |
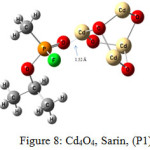 |
Figure 8: Cd4O4, Sarin, (P1) Click here to View figure |
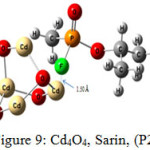 |
Figure 9: Cd4O4, Sarin, (P2) Click here to View figure |
Increasing of bond length of O=P of Sarin at P1 complexes of ZnO (1.61 Å) and Zn4O4 (1.68 Å) in comparison to a single Sarin (1.57 Å) is related to interaction of oxygen (in P=O group) with Zn of ZnnOn (n=1, 4) of these complexes and approximately no changes have been observed for this bond length at Zn4O4, Sarin P2 and P1 and P2 complexes of Cd4O4, Sarin. The value of bond length in evaluation of interaction strength is not enough. Therefore, we must investigate other factors. Dipole moment gives clear information about the arrangement of charges in nanostructures. The result of dipole moment may indicate that the size effect of metal oxide is important and asymmetry in charge distribution in M4O4 nanocones can further be explained to achieve different electronic properties. At all of the structures, the dipole moment of connected structures are increased and dipole moment of P1 complexes are more than P2. We have investigated the electronic properties by natural bond orbital (NBO) analysis.
Also, the NBO analysis shows that natural charge of oxygen in P=O group and metal changes more at P1 complexes than the P2.
Since the increase of electron transfer is occurred with the decrease of the energetic difference between highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), the selection of metal oxide with for adsorption of types of gases becomes possible.
Table 2: Calculated ΔE int (Kcal/mol), bond lengths(Å), Dipole moment (Debye), Natural Charge of oxygen (Oxygen in phosphonyl group) and Natural Charge of metal (M) at B3LYP/6-31++G (d,p)
|
structure |
ΔE int
|
r (O=P)/ Å |
r(O…M)a r(F…M)b / Å |
μ / Debye |
Charge of Oxygen
|
Charge of M M= Zn or Cd |
|
Sarin |
– |
1.57 |
– |
3.3220 |
-0.99198 |
– |
|
ZnO |
– |
– |
1.67 |
5.6271 |
– |
1.18435 |
|
ZnO, Sarin, (P1) |
-19.73 |
1.61 |
2.02a |
7.1779 |
-0.99802 |
1.34649 |
|
ZnO, Sarin, (P2) |
-9.31 |
1.45 |
2.09b |
3.6821 |
-0.54000 |
1.08000 |
|
Zn4O4 |
– |
– |
1.92 |
8.1281 |
– |
1.41800 |
|
Zn4O4, Sarin,(P1) |
-35.06 |
1.68 |
1.28a |
14.7986 |
-1.19811 |
1.55882 |
|
Zn4O4, Sarin,(P2) |
-21.23 |
1.57 |
2.50b |
11.7991 |
-0.99779 |
1.37265 |
|
CdO |
– |
– |
1.955 |
6.2341 |
– |
0.95450 |
|
CdO, Sarin, (P1) |
-15.03 |
1.49 |
2.27a |
13.7927 |
-1.14511 |
1.13887 |
|
CdO, Sarin, (P2) |
-8.81 |
1.47 |
3.32b |
5.7354 |
-1.04373 |
1.02968 |
|
Cd4O4 |
– |
– |
2.18 |
7.7090 |
– |
1.32500 |
|
Cd4O4,Sarin,(P1) |
-37.11 |
1.57 |
1.52a |
14.3985 |
-1.17329 |
1.59929 |
|
Cd4O4,Sarin,(P2) |
-24.50 |
1.57 |
1.50b |
8.6060 |
-1.01324 |
1.51529 |
The electron transition is a factor of strength of interaction. Thus, the narrow band gap between the HOMO and LUMO levels results in easy transition of electrons from HOMO level to LUMO. Table 3 shows HOMO and LUMO energy gaps (Egap) for complexes. The calculated gap of energy for the bare ZnO is 2.39 eV and this decrease to 2.23 eV for bare Zn4O4. The Egap of Sarin, ZnO complexes have distinctively changed compared to that of the mere ZnO and is increased from 2.39 to 3.29 by 37.65 % change for P1and 3.13 eV by 30.96 % change for P2. It indicates that the electronic property of the ZnO is very sensitive to Sarin adsorption.
It is important to pay attention that the gap of HOMO-LUMO energy for CdO and Cd4O4 are same and equal to 2.04 eV. Also, a small change can be seen in Egap of Sarin, CdO complexes, about 29.41% in P1 and 30.39% in P2 complexes. By comparing Egap of MO (M= Zn or Cd) complexes, it is clear that change of Egap for P1 complex of ZnO is more than CdO complex and P2 complexes of MO.
Table 3: Calculated HOMO energies (EHOMO), LUMO energies(ELUMO), HOMO–LUMO energy gap (Egap) of pristine and Sarin adsorbed
|
Structure |
EHOMO |
ELUMO |
Egap |
%∆Egap eV |
|
eV |
||||
|
Sarin |
-7.75 |
0.51 |
8.26 |
– |
|
ZnO |
-6.96 |
-4.57 |
2.39 ev |
– |
|
Zn4O4 |
-6.09 |
-3.86 |
2.23 ev |
– |
|
ZnO,Sarin, (P1) |
-5.55 |
-2.25 |
3.3 |
37.65 |
|
ZnO,Sarin, (P2) |
-5.9 |
-2.77 |
3.13 |
30.96 |
|
Zn4O4,Sarin, (P1) |
-5.03 |
-2.72 |
2.31 |
4.93 |
|
Zn4O4,Sarin, (P2) |
-4.38 |
-2.09 |
2.28 |
2.24 |
|
CdO |
-6.26 |
-4.21 |
2.04 |
– |
|
Cd4O4 |
-5.9 |
-3.86 |
2.04 |
– |
|
CdO, Sarin,( P1) |
-4.54 |
-1.9 |
2.64 |
29.41 |
|
CdO, Sarin,(P2) |
-5.52 |
-2.85 |
2.67 |
30.39 |
|
Cd4O4,Sarin, (P1) |
-4.98 |
-2.55 |
2.43 |
18.62 |
|
Cd4O4,Sarin, (P2) |
-5.11 |
-2.91 |
2.2 |
52.82 |
The obtained results show that by varying size of metal oxide mentioned in Table 3, the interaction abilities changes.By evaluating HOMO/LUMO energy gaps, it is obvious that the energy gap of M4O4 complexes is lower than MO complexes. Therefore, electron transfer is more probable in the M4O4 complexes. %∆Egap of Zn4O4 complexes at both of the P1 (4.93%) and P1(2.24%) positions are lower than Cd4O4 (18.62% for P1 and 52.82% for P2) complexes. Therefore it seems like that nano metal oxide of Cd4O4 has more ability for interaction to Sarin than Zn4O4. Also, by comparing P1 and P2 complexes of Cd4O4 it is seen that adsorption of Sarin on the Cd4O4 at P2 position is more probable.
As pointed in Table 4, the charge transfer (QT) of P1 complex of ZnO (0.0194) is more than CdO (0.0142). This result is related to strong interaction of ZnO with Sarin at P1 position. As seen in the figure 1, the bond of propyl is fractured due to interaction of ZnO with Sarin, and this nerve agent is decomposed. In this work, QT of MO metal oxides of Zn and Cd at P2 position is approximately equal. The charge transfer of P1 and P2 complexes of Cd4O4 with 0.071 and 0.0039, respectively, show that in M4O4 form of nano metal, the interaction of Cd4O4 with Sarin is more probable than Zn4O4. These results have compatibility with %∆ Egap.
The results of quantum calculations show that for decomposition of Sarin, the ZnO and Cd4O4 are better structures, that ZnO affects on fraction of propyl bond (P1 position) and Cd4O4 interacts with Fluorine atom of Sarin (P2 position).
To understand the magnitude of Sarin adsorption on the surface of MnOn (M= Zn ,Cd and n=1,4), we have developed the study of Radial Distribution Function( RDF) parameter of Sarin under different temperatures 273, 373,473, 573 and 673 K at 1 atmosphere of pressure, using Forcite calculations by Materials Studio software [27]. We have considered a cubic simulation box (59.4 Å, 59.4 Å, 59.4 Å) that contains MnOn (M=Zn or Cd and n=1,4) structure and Sarin molecules which are quantity wise 5 times more than MnOn, Fig 10, and evaluated sum of Van der waals energies of optimized structures at P1 position of ZnnOn (n=1,4) and P2 position of CdnOn (n=1,4).
 |
Figure 10: cubic simulation box contains MnOn and Sarin molecules |
Figure 11 shows that Sarin is decomposed at 380 K due to stocky connection with ZnO. In other words, ZnO connects with Sarin by formation of covalent bond. The Van der waals energies of Zn4O4, CdO and Cd4O4 complexes are weaker, respectively. It can be seen in chart 1 that optimum temperature for interaction of these complexes are 320, 380 and 308 K, respectively.
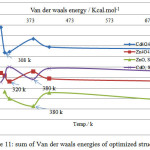 |
Figure 11: sum of Van der waals energies of optimized structures
|
Table 4: Charge transfer of Donor and Acceptor bonds for complexes at connected positions
|
Donor NBO (i)→ Acceptor NBO (j) |
E(2) kcal/mol |
Charge transfer QT |
|
ZnO, Sarin, (P1) LP (1) O11 →BD*( 1)Zn19 – O20 |
4.45 |
0.0194 |
|
ZnO, Sarin, (P2) LP (1) F13→BD*(1) O19 -Zn20 |
3.06 |
0.0048 |
|
Zn4O4, Sarin, (P1) LP (1) O18→ BD*(1) O19 -Zn20 |
22.91 |
0.0365 |
|
Zn4O4 ,Sarin, (P2) LP (1)F15→ BD*( 1) Zn3 – O6 |
5.15 |
0.0036 |
|
CdO, Sarin, (P1) LP (1) O14→BD*( 1)Cd19 -O20 |
9.16 |
0.0142 |
|
CdO, Sarin, (P2) LP (1) F13→ BD*(1 )Cd19 – O20 |
3.00 |
0.0053 |
|
Cd4O4,Sarin,(P1) LP (1) O18→ BD*(1)O19 -Cd23 |
52.37 |
0.0710 |
|
Cd4O4,Sarin, (P2) LP (1) F 13→ BD*(1) O15 -Cd22 |
1.81 |
0.0039 |
Conclusions
In this research, the study of decomposition adsorption of Sarin on ZnnOn and CdnOn (n=1,4) by B3lyp/6-31++g(d,p) quantum calculations shows that the ZnO can decompose Sarin by disconnection of propyl group of Sarin and formation of covalent bond with Sarin. Also, interaction of Cd4O4 with Fluorine of Sarin is important. The calculations of Radial Distribution Function support the quantum analysis results. The results of RDF show that the interaction of Sarin is probable at 380 K with ZnO and at 308 K with Cd4O4.
Acknowledgment
Thanks to Islamic Azad University, Yadegar – e- Imam Khomeini (RAH) Shahre Rey Branch, Islamic Azad University, Tehran, Iran, for funding this research project.
References
- Ganesan, K.; Reza, S.K.; Vijayaraghavan, R. J Pharm Bioallied Sci. 2010, 2, 166-178.
CrossRef - Degenhardt, C. E.A.M.; Pleijsier, K.; van der Schans, M. J. Journal of Analytical Toxicology. 2004, 28, 364-371.
CrossRef - Mawhinney, D. B.; Rossin, J. A.; Gehart, K.; Yates J. T. Jr. Langmuir. 1999, 15, 4789-4795.
CrossRef - Wagner, G.W.; Bartram, P.W. J MOL CAT A. 1999,144, 419-424.
CrossRef - Ekerdt, J. G.; Klabunde, K. J.; Shapley, J. R., White, J. M.; Yates, J. T. Jr. J. Phys. Chem. 1988, 92, 6182-6188.
CrossRef - Bermudez, V. M. J. Phys. Chem. C. 2007,111, 3719-3728.
- Bermudez, V. M. J. Phys. Chem. C. 2009,113, 1917-1930.
- Michalkova, A.; Ilchenko, M.; Gorb, L.; Leszczynski, J. J. Phys. Chem. B. 2004,108, 5294-5303.
CrossRef - Bermudez V.M. J Phys. Chem. C. 2007,111, 9314-9323.
- Rusu, C. N.; Yates, J. T. J. Phys. Chem. B. 2000, 104,12292-12298.
CrossRef - Paukku Y.; Michalkova, A., Leszczynski, J. J. Phys. Chem. C. 2009, 113, 1474-1485.
- Oha, S.W.; Kima, Y.H.; Yoob, D.J.; Oha, S.M.; Parkb, S.J. Sens. Actuat. B. 1993,13, 400-403.
- Sharma, N.; Kakkar, R. ADVANCED MATERIALS Letters. 2013, 4, 508-521.
- Adhikari, K.; Ray, A. K. J Nanopart Res. 2012, 14, 816.
- Iijima, S.; Ichihashi, T.; Ando Y. Nature. 1992, 356, 776-778.
- Alfieri, G.; Kimoto, T. Appl Phys Lett. 2011, 98, 123102.
- Ge, M; Sattler, K. Chem. Phys. Lett. 1994. 220,192-196.
- Chang, J.; Waclawik, E. R. J Nanoparticle Research. 2012,14,1012.
- Bagheri Ghomi, A. Journal of Structural Chemistry. 2016, 57, 194-198.
- Muscat, J.; Wander, A.; Harrison, NM. Chem Phys Lett. 2001, 342, 397-401.
CrossRef - Andersson, M. P.; Uvdal, P. J. Phys. Chem. A. 2005,109, 2937-2940.
CrossRef - Suenram, R. D.; Lovas, F. J. J. Mol. Spectrosc. 2002, 211,110-116.
CrossRef - Walker, A. R. H.; Suenram, R. D.; Samuels, A.; Jensen, J.; Ellzy, M. W.; Lochner, J. M.; Zeroka, D. J. Mol. Spectrosc. 2001, 207, 77-82.
CrossRef - Frisch MJ, T. G. Gaussian Inc., Wallingford, Revision A.1. edn. 2009.
- Nanotube Modeler JCrystal Soft. 2014 software.
- Nagarajan, V.; Chandiramouli, R.; Sriram, S.; Gopinath, P. J Nanostruct Chem. 2014, 4, 87-102.
CrossRef - Materials Studio v.6.0.0. 2011 software.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


