Synthesis, Spectroscopic Characterization, Computational Exploration of 6-(2-(2, 4-Dinitrophenylhydrazano)-Tetrahydro-2 Thioxopyrimidin-4(1h)-one
N. Kalaiarasi and S. Manivarman
Department of Chemistry, Government Arts College, C-Mutlur, Chidambaram-608102, Tamil Nadu, India.
Corresponding Author E-mail: drsmgac@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/330136
The structural and vibrational properties of 6-(2-(2,4-dinitrophenylhydrazano)-tetrahydro-2-thioxopyrimidin-4(1H)-one (3) prepared by condensation of synthesized thiobarbituric acid (1) with 2,4-dinitrophenylhydrazine (2) were studied using experimental FT-IR, FT-Raman, NMR spectra and theoretical calculations based on the density functional method were the simulated spectra coincides with experimental spectra. By means of HOMO-LUMO analysis charge transfer and stability of the molecule is analyzed. The stability of the molecule arising from hyper-conjugative interaction and charge delocalization has been analyzed using NBO analysis. Molecular electrostatic Potential was performed by B3LYP/6-31G (d, p) method. The atomic charge calculated by NBO is compared with Mulliken net charge. First hyperpolarizability is calculated in order to find its role in non-linear optics. Thermodynamical parameters entropy, enthalpy and heat capacities are calculated according to temperature changes involved.
KEYWORDS:DNPHTTO (3); FT-IR; FT-Raman; NBO; HOMO-LUMO; B3LYP.
Download this article as:| Copy the following to cite this article: Kalaiarasi N, Manivarman S. Synthesis, Spectroscopic Characterization, Computational Exploration of 6-(2-(2, 4-Dinitrophenylhydrazano)-Tetrahydro-2-Thioxopyrimidin-4(1h)-one. Orient J Chem 2017;33(1). |
| Copy the following to cite this URL: Kalaiarasi N, Manivarman S. Synthesis, Spectroscopic Characterization, Computational Exploration of 6-(2-(2, 4-Dinitrophenylhydrazano)-Tetrahydro-2-Thioxopyrimidin-4(1h)-one. Orient J Chem 2017;33(1). Available from: http://www.orientjchem.org/?p=30291 |
Introduction
In organic materials, hydrazones and its derivatives attracted attention due to crystallizing nature of asymmetric structure and flexibility that offer modification of non-linear properties. The nonlinear optical properties were investigated using nanosecond and femto second laser pulses [1]. Nonlinearities in hydrazones finds its potential application in all optical switching, data processing and eye and sensor protection [2]. Non – Analytical applications of their complexes are used in making rubber, as photographic materials, as dyes for wool and in industry [3-4]. In synthetic chemistry they are used as indicators, photo-thermochromic, sterilizers in houseflies and precursors [5]. Jiang discussed some of the analytical applications like transition metal binders [6]. Some of the important industrial applications were discussed by Sheldrick and John such as plasticizers, stabilizers for polymers as polymerization initiators, catalytic processes, and in waste water treatment [7-8].
Literature survey reveals that quantum calculations for title compound have not yet been reported so far. The main objective is to give a complete description about molecular geometry and molecular vibrations of title compound. For that purpose, quantum chemical computations were carried out on 6-(2-(2, 4-Dinitrophenyl hydrazano)–Tetrahydro – 2 – Thioxopyrimidin – 4 (1H) – One (3) using Density Functional Theory (DFT) and Total Energy Distribution has been observed. In addition, natural bonding orbitals analysis was also carried out in order to study any intra molecular charge transfer within the molecule.
Experimental
Synthesis of Thiobarbituric Acid
About 6g (0.25mol) of sodium metal is dissolved in 200 mL of ethanol. To this solution 15g (0.25mol) of thiourea and 40 mL of diethyl malonate is added. The reaction mixture is refluxed for 6 hrs in an oil bath and then vacuum distillated to make ethanol recovery. The clear solution thus obtained is filtered, cooled in ice bath overnight and the resulting solution is acidified with HCl. The crude product obtained is collected, washed with 50 mL water and dried in oven at 105°C-110°C for nearly 4 hrs. The obtained compound Thiobarbituric acid (TBA) is purified by recrystallization with ethanol and their respective melting point is about 243°C with 80% yield is obtained.
Synthesis of 6-(2-(2, 4-Dinitrophenyl hydrazano) – Tetrahydro – 2 –Thioxopyrimidin – 4 (1H) – One
Equimolar amount of thiobarbituric acid 2.44g (0.1mol) and 2,4-dinitrophenyl hydrazine 2.08g (0.1mol) are dissolved separately with 20 mL of ethanol, both the contents are mixed well until we gets a clear solution, then it is refluxed constantly for 4 hours andallowed to stand overnight. The product precipitated is separated by filtration, washed with ethanol, and air dried. The dark reddish coloured powder product obtained is recrystallized for further purification and the melting point is found to be 1800°C, with yield of about 67%. The schematic reaction and mechanism are shown in scheme 1 and scheme 2.
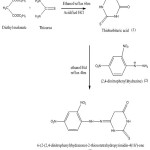 |
Scheme 1: synthesis of DNPHTTO by substitution-elimination reaction. Click here to View Scheme |
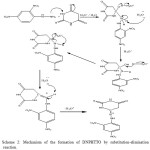 |
Scheme 2: Mechanism of the formation of DNPHTTO by substitution-elimination reaction. Click here to View Scheme |
Results and Discussion
Structural Determination by 1H NMR and 13C NMR Spectra
6- (2 – (2, 4 –Dinitrophenyl) Hydrazono)-Tetrahydro-2-Thioxopyrimidin-4(1h)-one
The 1H NMR spectrum of DNPHTTO was recorded in DMSO-d6 solvent. There is a singlet at δ 1.321 ppm integrated to 2H which should be the CH2 methylene proton. The NH protons exhibited two characteristic Broad singlets at δ 11.087 ppm and 8.775 ppm are assigned to NH of thioamide and amide in thiobarbituric acid. A medium peak at δ 8.541 ppm is the characteristic NH peak attached to aromatic ring. The signals of aromatic CH protons are observed as a multiplet at δ 7.25-7.30, 7.42, 7.55 ppm respectievely.
The 13C NMR spectra of DNPHTTO (DMSO-d6) are assigned as follows where the aromatic carbon shows signals at δ 150, 144.54, 131.97 ppm. The C=N hydrazone linkage is observed at δ 168.38 ppm, where the C-N Phenyl linkage is assigned at δ 163.20 ppm. The CH2 (methylene) carbon is assigned at δ 71.84 ppm, the blue shift is observed due to the substitution of phenyl ring where intra-molecular interaction and conjugation of lone pair of electrons between two rings occurs. The C=O and C=S functional group are observed at 172.67 ppm and 175.67 ppm.
Computational Chemistry
Molecular electronic structure has arrived an enormous amount of awareness by chemists and has become a key stone of modern chemical research. Theoretical calculations are used to optimized the geometry and vibrational wavenumbers with the Gaussian 03 program package using DFT (B3LYP/6-31G (d, p) basis sets. The assignments have been made on the basis of the corresponding PED analysis using Vibrational Energy distribution Analysis VEDA program [9-10]. To improve the agreement between the predicted and observed frequencies, the unscaled frequencies obtained are usually scaled for comparison.
Molecular Geometry
The optimized structure parameters of DNPHTTO calculated by B3LYP ab initio (B3LYP/6-31 G(d,p) basis set are listed in Table S1(BOND PARAMETERS OF DNPHTTO). In this step, all the calculations were performed using Gaussian03w program package [11] with the default convergence criteria without any constraint on the geometry [12].
 |
Table S1: Bond Parameters Of Dnphtto |
The title molecule Fig 1 shows variation in bond distance and bond length due to substituents attached to it. From the theoretical results, the aromatic ring is distorted from regular hexagon due to steric effect of the –NO2 group. The N-O bond distance at C3 and C5 position are 1.2308 A°, 1.2322 A°, 1.2256 A° whereas the N13-O14 bond distance is slighty higher compared with other N-O values may be due to the substitution of C=N at the neighbouring atom C6 . C=O bond distance is observed at 1.266 A° in literature [13] were in our title compound the C22=O25 bond distance is observed at 1.2113 A°. Theoretically predicted bond lengths of functional groups C-H, N-H are long. Generally the C-N bond is observed at 1.47 A° and C=N bond length is observed at 1.33 A°, where the C-N values are higher than C=N bond length [14]. The C-N bond length of phenyl ring is computed at 1.3645 A°, 1.4538 A°, 1.3645 A° for C3-N10, C5-N13, C6-N16 and for C-N of thiobarbituric ring the C22-N24, C21-N24, C21-N23, C19-N23 is observed at 1.3958 A°, 1.3794 A°, 1.3777 A°,
1.394 A°. Further, the phenyl ring C-N is higher than thiobarbituric ring C-N may be due to resonating structure of phenyl ring when compared with the heterocyclic ring. The hydrazone formation of our compound C19=N17 bond angle is observed at 1.3645 A° [15]. The C-C and C-C-C bands in the six membered rings are similar to the benzene molecule. The C-C and C-H bond lengths of DNPHTTO are found coincide with literature [16-20]. The DFT calculations give shortening of the angles at C1-C6-C5 by 2.7°, C5-C4-C3 by 0.7°, C3-C2-C1 by 0.3° and enlargement in angles at C6-C5-C4, C4-C3-C2 by 1.1° and C2-C1-C6 by 1.2° from normal angle 120° for benzene, which reveals that the substitution of NO2 group at C5 and C3 position causes this distortion. And for N23-C19-N17 and N17-C19-C20 are calculated at 125° and 119° at C19 position the difference observed may be due to the presence of lone pair of electrons at N23 position and active hydrogen at C20 position. The bond angles of the NO2 group of the title compound O15-N13-O14 (122°) , O11-N10-O12 (124°), O15-N13-C5 (118°), O12-N10-C3 (117°) are in agreement with the value 123.5°, 118.7°, 117.9° [21] also for N16-N17=C19, N23-C21=S26, N24-C22=O25 the trihedral angles are reported at 117°, 122°, 123°.
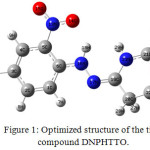 |
Figure 1: Optimized structure of the title compound DNPHTTO. Click here to View Figure |
Vibrational Assignment
The fundamental vibrations of a non-linear molecule consists of N atoms equal to (3N-6), apart from three traslational and three rotational degrees of freedom [22,23]. Table S2(VIBRATIONAL ASSIGNMENTS OF DNPHTTO) present the calculated vibrational frequencies and experimental values. The DNPHTTO molecules have 30 atoms with 84 normal vibrations which are distributed among the molecule considering Cs symmetry. All the 84 modes of vibrations are distributed as 29 stretching, 55 bending (in-planes, torsional and out of plane) vibrations in the infrared spectrum. The Cs structure was the lowest in energy at all levels. Therefore, we calculated the Cs symmetry of the molecule. The vibrational assignments are made by illustration of modes animated using the Gauss view program and are also justified with the help of TED analysis. The combined IR and Raman vibrational spectra of DNPHTTO are shown in Fig 2.
 |
Table S2: Vibrational Assignments Of Dnphtto |
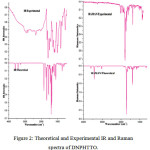 |
Figure 2: Theoretical and Experimental IR and Raman spectra of DNPHTTO. Click here to View Figure |
Methylene Vibration
The CH2 group has 6 vibrational modes asymmetrical stretching, symmetrical stretching, scissoring, wagging, twisting, rocking. The asymmetrical stretching of methylene groups ν as CH2, symmetric νs CH2, scissoring δ CH2, wagging vibration ω CH2 appear in the range 2980-3020, 2875-2925, 1430-1450, 1315-1365 cm-1 respectievely [24,25]. In our title compound the CH2 stretching modes are observed at
For the title compound, the CH2 stretching modes (asymmetric and symmetric modes) are observed at 2924 cm-1 in IR spectrum and 3086 cm-1 in Raman spectrum respectively and the symmetric mode is detected at 283 cm-1 in IR and 2972 cm-1 in Raman spectrum and they well coincides with the scaled wave numbers 3043cm-1, 2917 cm-1 with mode nos.77, 78 (TED = 96%) respectievely. The CH2 scissoring modes are observed at 1409 cm-1 in Raman Spectrum which agrees well with scaled wave nos. 1409 cm-1 with TED of about 86% (mode no. 67).
Aromatic Ring Vibration
The aromatic ring vibrational modes of title compound are analyzed with the help of previously published vibrations of the benzene molecule which is helpful in the identification of the phenyl ring modes [26]. The ring stretching vibrations are very prominent, as the double bond is in conjugation. Benzene ring possesses six ring stretching vibrations, of which the four with the highest wavenumbers (occurring near 1600, 1580, 1490 and 1440 cm-1) are good group vibrations. With heavy substituent, the bands tend to shift to somewhat lower wavenumbers. In aromatic ring compound, the C-H stretching vibrations are usually observed below 3000cm-1 [27-28]. In our title compound the CH stretching vibration is experimentally observed at 3118 cm-1 in FT-IR and 3268 cm-1 in FT-Raman at an intensity of 96% in TED with respective (mode no.81) and the experimental values are well coincides with the theoretical scaled value at 3144 cm-1 obtained by 6-31G(d,p) method.
C=S Vibration
The C=S group is less polar than the C=O group and has a considerably weak band. Compound that contains a thiocarbonyl groups, which shows absorption in the region 1250-1020 cm-1 [20]. In our present study, the C=S stretching frequency is observed in the region 1122 cm-1 and 1124 cm-1 in IR and FT-Raman spectra. The above experimental values are well coincides with the scaled theoretical value 1125 cm-1 obtained by B3LYP method.
C=O Vibration
The interaction of carbonyl group with neighbouring atoms did not creates a characteristics changes in C=O stretching frequency as did by N-H stretch. The C=O bond is formed by π-π interaction and due to different electro-negativities the bonding electrons are not equally distributed between the two atoms. The lone pair of electrons on oxygen also determines the nature of the carbonyl group. The position of C=O stretching vibration is very sensitive to various factors such as physical state, electronic effects by substituents, ring strains [29-31] etc. Normally C=O stretching vibrations is observed at 1850-1600 cm-1[32]. In our present study the wavenumbers observed at 1653 cm-1 in Raman are assigned to C=O stretching vibrations and it agrees with the computed values 1767 cm-1.
C=N vibration
The C=N stretching vibrational bands often mix with C=C vibrational bands and identification of such bands is too weak unless there are other substituent groups, thus the reliability of C=N assignment in IR spectrum is difficult; however there is a strong absorption band in Raman Spectrum. For this reason Raman Spectroscopy is often taken as reliable method to analyze C=N vibrations. From the literature, the C=N stretching vibration fall in the range 1535-1666 cm-1 [33-34]. The C=N stretching mode was experimentally observed at 1421.54 cm-1 in IR and 1427cm-1 in Raman in our present study for mode no.68 with TED (26 %) and the values are in good agreement with the scaled wave nos. 1430 cm-1.
NO2 vibration
The characteristic group frequencies of the nitro group are relatively independent of the rest of the molecule which makes this group convenient to identify. For the assignments of NO2 group frequencies basically six fundamentals can be associated to each NO2 groups namely, NO2 symmetric stretching, NO2 Asymmetric stretching, Scissoring, rocking (In plane vibrations). In addition to NO2 wagging, twisting modes of NO2 group would be expected to be depolarized for out of plane symmetry species. The NO2 asymmetric stretching vibration band range is 1625-1540 cm-1, and symmetric stretching vibration [35] is 1400-1360 cm-1. The NO2 asymmetric stretching vibration is experimentally observed in the range 1593 cm-1 in IR and 1600 cm-1 in Raman is assigned to asymmetric stretching vibration. The band established at 1292 cm-1 in IR and 1322 cm-1 in Raman is assigned to symmetric stretching vibration coincides with theoretical wavenumber 1336 cm-1 and 1303 cm-1 (mode no. 62 (31%) & 60 (17 %)). Further Aromatic Nitro Compounds have a band of weak to medium intensity in the region 590-500 cm-1 due to in plane deformation mode of the NO2 group. Probably in our title compound the respective peak is observed at 812cm-1 in IR and at 805cm-1 in Raman spectra with TED (32%) and mode no.40 and it coincides well with the scaled values 810cm-1.
N-H vibration
In heterocyclic compounds, the N-H stretching vibrations generally occur in the region 3500-3000 cm-1 [36]. The position of absorption in this region depends upon the degree of hydrogen bonding, and hence upon the physical state of the sample. The spectral lines assigned to N-H stretching vibrations have shifted to higher region in the present system. It clearly indicates that the stretching of N-H bond upon protonation has shifted the frequency to a higher region. In our title molecule, the N-H stretching vibration is predicted at 3292 cm-1 in IR and 3320 cm-1 in Raman by B3LYP / 6-31G (d, p) method which matches with the scaled wave numbers 3300 cm-1 with TED (82%) and respective mode at 82.
In order to investigate the performance and vibrational wavenumbers of the title compound root mean square value (RMS) and correlation coefficient between calculated and observed wavenumbers were calculated and correlation graph is represented in Fig 3.
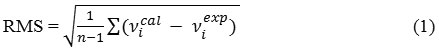
RMS values of wavenumbers were evaluated using the following expression [37]. The RMS error of the observed IR and Raman bands are found to be 18.8, 12.67 for DFT method. The small difference is observed since experimental vibrational modes are calculated in solid phase and Scaled vibrational modes are calculated in gaseous phase.
 |
Figure 3: Correlation graph of Theoretical value Vs Experimental value of DNPHTTO. Click here to View Figure |
Non-linear optical properties
The significance of the polarizability and the first hyperpolarizability of molecular systems depend on interaction between acceptor and the donor groups, since it is the base to intramolecular charge transfer [38]. The charge transfers through single-double bond conjugated path induce large variations in dipole moment and molecular polarizability whereas it is strong at IR and Raman activity. Hydrogen bond interactions enhance molecular hyperpolarizability. Dipole moment (μ), polarizability (α), hyperpolarizability (β) calculated were 1.9252 Debye, 5.3048 x10-30esu and 24.2991 x10-30esu (β value obtained is 14 times higher than the standard β0 value of urea) Table S3 (NLO PARAMETRS OF DNPHTTO).
Table S3: Nlo Parametrs of Dnphtto
|
Parameters |
B3LYP/6-31G(d,p) |
|
Dipole moment ( μ ) Debye |
|
| μx | 1.3904 |
| μy | -1.2664 |
| μz | -0.4119 |
| μ | 1.9252Debye |
| Polarizability ( α0 ) x10-30esu | |
| αxx | 362.15 |
| αxy | 18.01 |
| αyy | 225.67 |
| αxz | -1.97 |
| αyz | -2.12 |
| αzz | 78.64 |
| α | 5.3048x10-30esu |
| Hyperpolarizability ( β0 ) x10-30esu | |
| βxxx | -2452.55 |
| βxxy | -445.12 |
| βxyy | -345.49 |
| βyyy | 177.69 |
| βxxz | -86.22 |
| βxyz | 64.49 |
| βyyz | 119.16 |
| βxzz | -2.30 |
| βyzz | 5.03 |
| βzzz | -25.11 |
| β0 |
24.2991x10-30esu |
Standard value for urea (μ=1.3732 Debye, β0=0.3728×10-30esu): esu-electrostatic unit
Electron Density and the Electrostatic Potential Surface
The molecular electrostatic potential surface (MESP) is a method of mapping electrostatic potential onto the iso-electron density surface simultaneously displays molecular shape, size and charge of molecules in three dimensional representations [39]. Molecular interaction is discussed to find the reactive site of the molecule. In MEP the Hydrogen atom are represented by blue region. The molecular electrostatic potential (MEP) at a point r around a molecule is expressed as
![]()
ZA – charge on nucleus A, V(r) – the resultant at each point r,
The first and second terms represent the contributions to the potential due to nuclei and electrons, respectively. The total electron density and MESP surfaces of the molecules under investigation are constructed by using B3LYP/6-31G (d, p) method. The electrostatic potential increases in the order red<orange<yellow<green<blue. where the colour scheme of MESP in the negative electrostatic potentials are shown in red, the intensity of which is proportional to the absolute value of the potential energy, and positive electrostatic potentials are shown in blue while Green indicates surface areas where the potentials are close to zero. Fig 4 shows the molecular electrostatic potential surface of DNPHTTO.
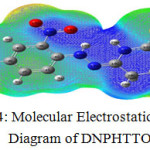 |
Figure 4: Molecular Electrostatic Potential Diagram of DNPHTTO. |
Local negative electrostatic potential (red) signal indicates in the O25 and N23 atoms whereas local positive electrostatic potentials (blue) signal indicates in the H18 of hydrazone linkage and thiobarbituric ring. Green areas cover parts of the molecule where electrostatic potentials are close to zero (C–C bonds) of phenyl ring. GAUSSVIEW visualization program has been utilized to construct the MESP surface.
Frontier Molecular Orbital Analysis
Frontier molecular orbital explains about energy transfer in the molecule. Energy of the HOMO is directly related to the ionization potential, LUMO is directly related to the electron affinity. The difference in energy between transition levels is called as energy gap, which is responsible for the stability of the structures [4041]. The HOMO – LUMO energy gap of DNPHTTO is about -3.4892 eV and HOMO-1 to LUMO+1energy gap is -4.1317 eV and represented diagrammatically Fig 5. The energy values of each transition levels are tabulated as Table S4 (HOMO-LUMO table of DNPHTTO). The Density spectrums of DNPHTTO Fig 6 were obtained by occupied and virtual frontier molecular orbital energies which supports FMO in order to find energy gap.
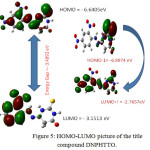 |
Figure 5: HOMO-LUMO picture of the title compound DNPHTTO. Click here to View Figure |
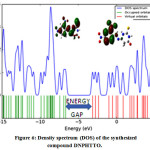 |
Figure 6: Density spectrum (DOS) of the synthesized compound DNPHTTO. Click here to View Figure |
UV spectral analysis
The UV-vis spectra provide evidence confirming their molecular structures by their maximum wavelength absorption. The λmax for lower lying singlet states of the molecule have been calculated by DFT/ B3LYP/ 6-31 G (d, p) method. The λmax, oscillator strength and excitation energies are reported in Table S5 (UV PARAMETERS OF DNPHTTO). Visible absorption maxima correspond to electronic transition between frontier orbitals is considered. The observed and calculated UV-Vis Spectra in gas phase are compared and illustrated in Fig 7 (a) and (b). The n-π* transition energy is calculated at 394.34 nm in gas phase, above the experimental maximum in methanol at 343.5 nm with strong intensity in solvent phase. The π-π* transition observed at 362.78 nm, in gas phase, and a weak peak is observed at 306.5 nm in the experimental maximum in methanol. Increase in polarity of the solvent generally shifts the n-π* and n- * bands to shorter wavelength and π-π* bands to longer wavelengths. The differences in experimental and theoretical values are due to solvent effect and resonating nature of aromatic ring and lone pair electrons.
Table S5: Uv Parameters of Dnphtto
|
Excitation |
Oscillator strength |
Energy (eV) |
Wavelength (nm) |
FORCE CONSTANT |
|
|
Theoretical gas phase value |
Experimental value |
||||
| Excited State 1: | Singlet-A | 3.0056 eV | 412.51 nm | f=0.1123 | |
| 83 -> 84 | 0.66179 | ||||
| Excited State 2: | Singlet-A | 3.1441 eV | 394.34 nm | 343.5nm | f=0.0012 |
| 82 -> 84 | 0.52024 | ||||
| 82 -> 85 | -0.30638 | ||||
| 82 -> 86 | 0.33675 | ||||
| Excited State 3: | Singlet-A | 3.4176 eV | 362.78 nm | 306.5nm | f=0.0057 |
| 82 -> 84 | 0.47249 | ||||
| 82 -> 85 | 0.36419 | ||||
| 82 -> 86 | -0.34915 | ||||
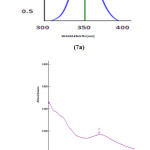 |
Figure 7: Theoretical (7a) and experimental (7b) UV spectra of the synthesized DNPHTTO compound. |
Global and Chemical Reactivity Descriptors
Based on the energies of frontier molecular orbital energies, various chemical reactivity descriptors as well as global reactivity descriptors have been defined [42].These parameters are used to find molecular stability, optical polarizability and reactivity of a compound.
Chemical hardness signifies resistance towards deformation or polarization of the electron cloud of the atom, ions or molecules under small perturbation of chemical reaction. Hard molecules have a large energy gap and soft molecules has small energy gap. Using Koopmans theorem for closed-shell compounds, η, μ and χ be defined as,
![]()
(where I= -EHOMO and A= -ELUMO)

Where A and I are the ionization potential and electron affinity of the compounds respectievely. Electron affinity refers to the capability of ligand to accept one electron from a donor. A new descriptor to quantify the global electrophilic power of the compound is electrophilicity index describes the biological activity respectievely. The usefulness of this new reactivity quantity (ω) has been recently demonstrated in understanding the toxicity of various pollutants in terms of their reactivity and site selectivity. [43-45]
![]()
The electronegativity χ (chi) of an element is the power of an atom of the element to attract electron to itself when it is part of a compound. If an atom has a strong tendency to acquire electron, it is said to be highly electronegative. Electronegativity is a very useful concept in chemistry and has numerous applications which include a rationalization of bond energies and the type of reaction that substance undergoes and the prediction of the polarity of bonds in molecules. It can be related to the size and electronic configuration of the atom. The properties of individual atom was proposed by Robert Mulliken i.e., an atom with high ionization energy (I) and a high electron affinity (Ea) likes to acquire electron than to lose electron when it is part of a compound, i.e highly electronegative. Conversely the atom with low ionization energy and electron affinity tends to lose electron rather than gain them i.e. electropositive. These observation motivated the definition of the mulliken electronegativity; χm as the average value of the Ionization energy and the electron affinity of the elements. From calculation the electronegativity (χm) of the compound is calculated.
![]()
By using above eqns. (3)-(7) the values of η, μ, S, χm and ω are determined and represented in Table 1. It is seen that the chemical potential of the title compound is negative and it means that the compound is stable. The hardness signifies the resistance towards the deformation of electron cloud of chemical systems under small perturbation encountered during the chemical process. Hard systems are relatively small and less polarizable.
 |
Table 1: Global and chemical parameters of DNPHTTO. |
Natural Bond Orbital Analysis
Natural bond orbital explains about hybridisation involved in bond orbitals. Interaction between atomic orbitals (filled donors) – Lewis type NBO’S and empty (acceptors) non-Lewis NBO’s are reported. The hyper-conjugation may be given as stabilizing effect that arises from an overlap between an occupied orbital with empty orbital, when the orbitals are properly oriented.
This non covalent bonding interaction can be quantitatively described in terms of the NBO analysis. Some electron donor orbital, acceptor orbital and the interacting stabilization energy resulting from second order microdisturbance theory is reported. The occupancy and energies are calculated by NBO analysis. The stabilization energy ΔEi,j associated with delocalization is estimated by the second order perturbative as

qi – donor orbital occupancy, εi , εj – diagonal elements (Energies of orbital), F (i ,j) – off diagonal NBO Fock or Kohn – Sham matrix element [46], E (2) – Stabilization Energy.
The intra-molecular interaction are formed by the orbital overlap between (c-c) and *(c-c), non bonded Nitrogen and Oxygen atoms with antibonded carbon, Nitrogen, Oxygen and Sulphur which leads to moderate stabilization energy. From Table S6 (Second order perturbation theory analysis of Fock matrix in NBO basis for DNPHTTO) the intra-molecular hyperconjugative interactions formed by orbital overlap π(c-c) π*(c-c) are calculated, these interactions are observed as an electron density in c-c antibonding orbital that weakens the respective bonds. The ED at the six conjugated π bonds
Table S6: Second Order Perturbation Theory Analysis of Fock Matrix in Nbo Basis for Dnphtto
|
Type |
Donor NBO (i) |
ED/e |
Acceptor NBO (j) |
ED/e
|
E(2)
KJ/mol |
E(2)
kcal/mol |
E(j)-E(i)
a.u. |
F(i,j)
a.u. |
|
π -π* |
C3 – C4 |
1.67180 |
C1 – C 2 |
0.24297 |
90.63 |
21.66 |
0.30 |
0.074 |
|
C 5 – C 6 |
0.49046 |
58.28 |
13.93 |
0.27 |
0.057 |
|||
|
N10 – O11 |
0.63682 |
103.64 |
24.77 |
0.16 |
0.061 |
|||
|
π -π* |
C5 – C6 |
1.59092 |
C 1 – C 2 |
0.24297 |
46.69 |
11.16 |
0.31 |
0.054 |
|
C 3 – C4 |
0.33407 |
107.40 |
25.67 |
0.30 |
0.079 |
|||
|
C 5 – C6 |
0.49046 |
6.44 |
1.54 |
0.27 |
0.019 |
|||
|
N13 – O15 |
0.62556 |
141.88 |
33.91 |
0.15 |
0.068 |
|||
|
π*-π* |
C5 – C6 |
0.49046 |
C 1 – C 2 |
0.24297 |
350.12 |
83.68 |
0.04 |
0.080 |
|
C3 – C4 |
0.33407 |
523.04 |
125.01 |
0.03 |
0.081 |
|||
|
n -σ* |
LP ( 2) O11 |
1.89587 |
C3 – N10 |
0.10340 |
53.60 |
12.81 |
0.57 |
0.077 |
|
N10 – O12 |
0.05696 |
80.71 |
19.29 |
0.70 |
0.105 |
|||
|
n -σ* |
LP ( 2) O12 |
1.89785 |
C 3 – N10 |
0.10340 |
52.09 |
12.45 |
0.57 |
0.076 |
|
N10 – O11 |
0.05644 |
79.96 |
19.11 |
0.71 |
0.105 |
|||
|
n -π* |
LP ( 3) O12 |
1.44896 |
N10 – O11 |
0.63682 |
678.23 |
162.10 |
0.14 |
0.138 |
|
n -π* |
LP ( 3) O14 |
1.51555 |
N13 – O15 |
0.62556 |
537.35 |
128.43 |
0.16 |
0.131 |
|
n -π* |
LP ( 1) N16 |
1.65136 |
C 5 – C 6 |
0.49046 |
197.19 |
47.13 |
0.28 |
0.107 |
|
N 16 – H 18 |
0.06719 |
5.40 |
1.29 |
0.72 |
0.029 |
|||
|
N 17 – C 19 |
0.26743 |
92.34 |
22.07 |
0.29 |
0.073 |
|||
|
n -σ* |
LP ( 1) N17 |
1.91867 |
N 16 – H 18 |
0.06719 |
27.87 |
6.66 |
0.83 |
0.066 |
|
C 19 – C 20 |
0.02740 |
6.57 |
1.57 |
0.78 |
0.032 |
|||
|
C 19 – N 23 |
0.05722 |
63.60 |
15.20 |
0.80 |
0.099 |
|||
|
n -σ* |
LP ( 1) N23 |
1.64869 |
N 17 – C 19 |
0.01580 |
4.60 |
1.10 |
0.88 |
0.030 |
|
N 17 – C 19 |
0.26743 |
169.33 |
40.47 |
0.29 |
0.098 |
|||
|
C 19 – C 20 |
0.02740 |
4.52 |
1.08 |
0.68 |
0.026 |
|||
|
C 21 – S 26 |
0.41305 |
270.16 |
64.57 |
0.22 |
0.107 |
|||
|
n -π* |
LP ( 1) N24 |
1.62967 |
C 21 – S 26 |
0.41305 |
278.28 |
66.51 |
0.21 |
0.106 |
|
C 22 – O 25 |
0.21686 |
202.97 |
48.51 |
0.29 |
0.110 |
|||
|
n -σ* |
LP ( 2) O 25 |
1.85360 |
C 20 – C 22 |
0.06509 |
89.75 |
21.45 |
0.62 |
0.105 |
|
C 22 – N 24 |
0.09303 |
123.09 |
29.42 |
0.67 |
0.127 |
E(2) means (stabilization energy).. Energy difference between donor(i) and acceptor(j) NBO orbitals. F(i,j) is the fork matrix element between i and j NBO orbitals.
(˜1.67180, 1.59092, 0.49046) to π*(˜0.63682, 0.62556, 0.33407 e) of the phenyl ring clearly demonstrates strong delocalization leading to stabilization of energy values as shown above. The most important interactions observed other than π- π* is n- π* and n- * were the E (2) value is O11 – (N10-O12), O12 – (N10-O11), N17 – (C19-N23), N23 – (C17-N19), O2 – (C22-N24) is 80.71, 79.96, 63.60, 169.33, 123.09 KJ / mol respectievely. And the interaction of lone pair of oxygen and nitrogen with nitrogen, oxygen and carbon is 678.23, 537.35 , 197.19, 278.28, 202.97 KJ / mol for transition between O12 – (N10-O11), O14 – (N13-O15 ), N16 – (C5 -C6), N24 – (C21-S26), N24 – (C22-O23 ). The increase in energy may be due to the presence of nitro group which results in delocalization of electron and hence decrease the stability of the molecule probably.
Thermodynamic properties
Scaling factors have been recommended [47] for an accurate prediction to determine the zero point vibrational energies for DFT calculation and it is found to be 127.823 K/ Cal/ mol. The total energy of the molecule is -1488.32 K/ Cal/ mol. In addition to this the dipole moment reflects the molecular charge and it is determined for neutral molecules Table S7
Table S7: Thermal Parameters of Dnphtto
|
Parameters B3LYP/ 6-31G(d,p) |
DNPHTTO |
|
Total Energies |
-1488.32 |
|
Zero point Energy |
127.823 |
|
Rotational constants |
0.54730 |
|
0.10794 |
|
|
0.09163 |
|
|
Entropy |
|
|
Total |
139.51 |
|
Translational |
0.8890 |
|
Rotational |
0.8890 |
|
Vibrational |
137.73 |
Thermal Parameters of DNPHTTO
On the basis of vibrational analysis at B3LYP/6-31 G (d, p) level, the standard statistical thermodynamic functions: heat capacity (C°p, m), entropy (S° m) and enthalpy changes (H° m) for the title compound were obtained from the theoretical
harmonic frequencies and are listed in Table S8 (Thermodynamic Parameters of DNPHTTO) . From Table S8, it can be observed that thermodynamic functions are increasing with Temperature ranging from 100-1000K due to the fact that the molecular vibrational intensities increase with temperature and it is graphically represented in the Fig 8.
Table S8: Thermodynamic Parameters of Dnphtto
|
T (K) |
S (J/mol.K) |
Cp (J/mol.K) |
ΔH (J/mol.K) |
| 100 | 274.82 | 33.26 | 3.33 |
| 200 | 297.88 | 33.26 | 6.65 |
| 298.15 | 311.16 | 33.26 | 9.92 |
| 300 | 311.36 | 33.26 | 9.98 |
| 400 | 320.93 | 33.27 | 13.30 |
| 500 | 328.36 | 33.32 | 16.63 |
| 600 | 334.44 | 33.47 | 19.97 |
| 700 | 339.62 | 33.75 | 23.33 |
| 800 | 344.15 | 34.13 | 26.72 |
| 900 | 348.20 | 34.62 | 30.16 |
| 1000 | 351.88 | 35.18 | 33.65 |
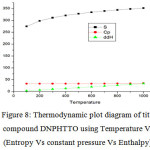 |
Figure 8: Thermodynamic plot diagram of title compound DNPHTTO using Temperature Vs (Entropy Vs constant pressure Vs Enthalpy). Click here to View Figure |
Conclusion
The observed FT-IR and FT-Raman values correlate well with considered wavenumbers. For accurate prediction of vibrational assignments PED were calculated using VEDA program. The NBO reveals hyperconjugative interaction and charge delocalization around the bonds. Charge transfer mainly preferred due to C6-N16 and C9-N17. The presence of C=N fused with the dintrophenylhydrazine and thioxopyrimidone ring, icreases the molecular hyperpolarizability of title compound about 24.229 x10-30esu , which is 14 times higher than that of urea. Stability and reactivity is analysed by energy gap of HOMO-LUMO. HOMO-LUMO are mainly located over N=C group. The global reactivity descriptors ω determines the biological activity nature of our compound and it is used to study about toxicity of various synthesized compounds. A thermodynamic calculation of the compound is calculated in gas phase and the thermodynamic parameters s, ΔE, ΔH finds to increase with increase in temperature and they could not be used in solution.
Reference
- Munn, R.W.; Ironside, C,N. Principle and applications of Nonlinear Optical Materials 1993.
- Vijayakumar, S.; Babu, M.; Balakrishna, K.; Chandra sekharan, K. Optik – International Journal for Light and Electron Optics 2012, 123, 21-25.
CrossRef - Lueda, H. Journal of the Chemical Society Abstracts 1986, 105, 32929.
- Buzykin, B, I.; Bystrykh, N.; Stolyarov, A. P.; Kitaev, K. Cheminform Abstracts 1978, 9, 4.
- Galiz J, C.; Rub J, C.; Edger, J. Nature 1955, 34, 176.
- Jiang, P.; Chen, L.; Lin, J.; Chemical Communications 2002, 13, 1424-1425.
CrossRef - Thom.; Sheldrick, G, M.; Acta Cryst. Section A Foundations of Crystallography 2008, 64, C221.
- John, R, P.; Sreekanth, A.; Maliyeckal, R.; Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2013, 59, 1349-1358.
CrossRef - Jamroz, M, H.; Spect. Acta Part A: Molecular and Biomolecular Spectroscopy 2013, 114, 220 – 230.
- Xiaosong Li.; Millam, J, M.; Scuseria, G, E.; Frisch, M. J.; Journal of chemical Physics 2003, 119, 7651.
CrossRef - Schlegel, H, B.; Journal of Computational Chemistry 1982, 3, 214-218.
CrossRef - Ricardo Infante Castillo.; Rivera – Montalvo.; Hernandez Rivera.; Journal of Molecular Structure 2008, 877, 10-19.
- Allen, F, H.; Acta Crystallographica Section A 1984, 40, 441.
CrossRef - Dalip Kumar.; Maruthi Kumar.; Soumitra Ghosh.; Kavita Shah.; Bioorganic & Medicinal Chemistry Letters 2012, 22 212 -215.
CrossRef - Golovnev, N, N.; Molokeev, M, S.; Tarasova, L, S.; Journal of Molecular Structure. 2014, 1068, 216-211.
CrossRef - Kunio Kohata.; Tsutomu Fukuyama.; Kozo Kuchitsu.; Journal of Physical Chemistry. 1982, 86, 602 – 606.
CrossRef - Naumov, V, A.; Litvinov, O, A.; Geise, H, J.; Journal of Molecular Structure 1983, 99 303-307.
CrossRef - Colthup, N, B.; Daly, L, H.; Preface. Introduction to Infrared and Raman Spectroscopy 1990, 11-12.
- Lide, D, R.; Tetrahedron 1962, 17, 125 – 134.
CrossRef - Kalaiarasi, N.; Manivarman, S.; International journal of science and technoledge 2016, 4, 181- 193.
- Iyengar, S.; Bernhard Schlegel, H.; Millam, J, M.; Journal of Chemical Physics 2001, 115, 10291.
CrossRef - Silverstein, R, M.; Bassler, G, C.; Morrill, T, C.; Magnetic Resonance in Chemistry 1992, 30, 364-364.
- Long, D, A.; Journal of Raman Spectroscopy 2001, 35, 905.
CrossRef - Gabor Pongor.; Geza Fogarasi.; Boggs.; Peter Pulay.; Journal of Molecular Spectroscopy 1985, 114, 445-453.
CrossRef - Varma, R, S.; Dahiya, R.; Tetrahedron 1998, 54, 6293-6298.
CrossRef - Jamroz, M, H.; Molecular and Biomolecular Spectroscopy 2013, 114, 220-230.
- Wong, K, C.; Journal of Chemical Education 2015, 92, 1602-1603.
CrossRef - Bakalska, R, I.; Delchev, V, B.; Journal of Molecular Modeling 2012, 18, 5133-5146.
CrossRef - Haiying Chen.; Qiheng Wang.; Yong Dai.; Chin Lin.; Superconductivity 2000, 337, 317-321.
CrossRef - Fabio Ramondo.; Andrea Pieretti.; Lorenzo Gontrani.; Chemical Physics 2001,271, 293 -308.
CrossRef - Eduardo Mendez.; Cerda.; Journal of Physical Chemistry 2007, 111, 3369-3383.
- Hajar Sahebalzamani.; Farshid Salimi.; Elmira Dornapour.; Journal of Chemistry 2013, 2013, 1-6.
- Daimay Lin-Vien.; Colthup, N, B.; Fateley, W, G.; Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules 1991, 73 – 94.
- Chisti, S.; Muchkund Dubey.; South Asian Survey 1997, 4, 186-188.
- Csizmadia, G.; International Journal of Quantum Chemistry 1976, 13, 159.
- Brodie, A, M.; Ranford J, D.; Inorganica Chimica Acta 1988, 267, 27-28.
- Onkar Prasad.; Leena Sinha.; Journal of molecular structure: Theochem 2010, 940 82-86.
CrossRef - Castiglioni.; Del Zoppo.; Journal of Raman Spectrosopy 1993, 24, 485-494.
- Tobias Beck.; Andrius Krasauskas.; Tim Gruene.; Acta Crystallographica Section D Biological Crystallography 2008, 64, 1179 – 1182.
CrossRef - Pearson, R, G.; National Academy of Sciences 1986, 83, 8440-8441.
- Gokhan Gece.; Corrosion Science 2008, 50, 2981-2992.
CrossRef - Lewis, D, F, V.; Ioannides, C.; Parke, D, V.; Xenobiotica 1994, 24, 401- 408.
CrossRef - Pratim Kumar Chattaraj.; Buddhadev Maiti.; Utpal Sarkar.; The Journal of Physical Chemistry A 2003, 107, 4973 – 4975.
- Subramani Saravanabhavan.; Palanisamy Thanikaivelan.; Environmental Science & Technology 2004, 38, 871 -879.
CrossRef - Mathammal, R.; Sangeetha, K.; Sangeetha, M.; Journal of Molecular Structure 2016, 1120, 1-14.
CrossRef - Kohn, W.; Sham, L.; Journal of physical Review A 1965, 140, 1133.
- Bevan Ott.; Juliana Boerio-Goates.; Statistical & Chemical Thermodynamics: Principles and Applications 2000, 497-592.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


