Effect of Platinum loading on Graphene Nano Sheets at Cathode
Rikson Siburian1, Kerista Sebayang2, Minto Supeno1 and Harlem Marpaung1
1Chemistry Department, Faculty of Mathematic and Natural Science,University of Sumatera Utara, Medan 20500, Indonesia.Nanomedicine Center-University of Sumatera Utara Stem cell- University of Sumatera Utara.
2Physic Department, Faculty of Mathematic and Natural Science, Universitas Sumatera Utara, Medan, Indonesia, 20500.
Corresponding Author E-mail: riksonsiburian2000@yahoo.com
DOI : http://dx.doi.org/10.13005/ojc/330114
The effect of Ptloading on graphenenanosheets (GNS) at electrode catalyst fuel cell was carried out. The purpose of this research is to know the effect of Pt loading on GNS and support material for catalytic activity of Pt/GNS. The results show 20 wt % Pt/GNS is highest catalytic activity among the others and thecatalytic activity of Pt on GNS is higherthan that of Pt on carbon black (CB) commercial catalyst. A core level shift of Pt 4f in XPS indicates that Pt is chemically interacted with GNS.It has been ascribed to the difference in the interface interaction between Pt and graphene via different strength of the π–d hybridization.
KEYWORDS:Pt; graphenenanosheet; Ptsubnano-clusters; ORR
Download this article as:| Copy the following to cite this article: Siburian R, Sebayang K, Supeno M, Marpaung H. Effect of Platinum loading on GrapheneNano Sheets at Cathode. Orient J Chem 2017;33(1). |
| Copy the following to cite this URL: Siburian R, Sebayang K, Supeno M, Marpaung H. Effect of Platinum loading on GrapheneNano Sheets at Cathode. Orient J Chem 2017;33(1). Available from: http://www.orientjchem.org/?p=29862 |
Introduction
Polymer electrolyte membrane fuel cells (PEMFC) are being developed as electrical power sources for vehicles and portable applications as an alternative to conventional internal combustion engines, secondary batteries, and other conventional power sources. Generally, the requirements for catalyst support materials can be summarized as: high specificsurface area, low combustive reactivity, high electrochemical stability, high conductivity, and interaction between catalytic metals and the support materials should be considered in improving the catalytic activity and durability.1
Recently, the design of cheap and stable fuel cell catalysts for catalyst electrode (oxygen reduction reaction (ORR)) at cathodeis main challenge.2Catalysts exhibit great influence on both the cost and the durability of PEMFC.3The platinum nanoparticles supported on carbon black (Pt/CB) are most used for ORR catalysts.It has outstanding catalytic and electrical properties and superior resistant characteristics to corrosion.4Therefore, much of the art and science of catalysts development for the ORR rely on both the fundamental understanding of the reaction at the Pt electrolyte interface and the optimization of the catalytic properties of the Pt surface.5However, Pt price is expensive and also it is limited natural resources.6So, it is a prerequisite to decrease the usage of Pt and enhance the catalytic activity of Pt in order to achieve a competitive low cost of fuel cell.
The catalyst support materials exhibit great influence on the cost, performance, and durability of PEMFC.For instance, carbon-supported precious metal nanoparticles (e.g., Pt, Au, Pd, and Rh) are widely used inheterogeneous catalysis and electrocatalysis.7The support materials are necessary to obtain a high dispersion, narrow distribution of Pt and Pt-alloy nanoparticles and also can interplay with catalytic metals, which is the prerequisite to obtain the high catalytic performance of catalysts.1Commonly, the catalyst support materials require high specific surface area, high conductivity, low combustive reactivity under both dry and humid air conditions at low temperatures (150 oCor less), high electrochemical stability under fuel cell operating conditions, easy to recover Pt in the used catalyst,1,8 and the interaction between catalytic metals and the support materials.9This is because the interaction between the support and metal catalyst can modify the electronic structure of catalytic metals which in turn changes the catalytic activity. 10.However, the weak interaction between metal and carbon supports results in a severe sintering/agglomeration of catalytic metal nanoparticles and consequently decreases the active surface area, which leads to the degradation of performance under long-term operations.7Many researchers have reported novel carbon support materials, such as carbon nanohorns,11carbon nanocoils,12carbon nanotubes (CNT),13graphite nanofibers (GNFs),14and carbon black15for PEMFC applications.Some papers also reported that nitrogen-doped carbon nanotubes (N-CNT) with metal catalysts or without metals on them show enhancement catalytic activity toward ORR.16Theyshowed promising results toward fuel cell electrode reactions: ORR and methanol oxidation reaction (MOR). But, in term of activity, cost and durability, current catalysts can still not satisfy the requirements of target PEMFC.17Therefore, many efforts are still necessary to find the novel catalytic metals and support materials.
Graphene sheets, a two-dimensional carbon material with single (or a few) atomic layer has attracted great attention for both fundamental science and applied research. This is caused it has large surface area (2630 m2 g-1),18and high carrier mobility (104 cm2 V-1 s-1 at room temperature).19Recently, grapheneas a supporting material for Ptcatalyst are believed to improve catalytic activity for hydrogen oxidation reaction (HOR) and methanol oxidation reaction (MOR). However, the controversial results regarding the ORR activity for Pt/GNS compare to Pt/CB commercial catalyst. Moreover, fuel cell tests with Pt/GNS catalysts as cathode materials showed a considerably lower performance than that of the cell with Pt/CB as cathode catalyst.20
In this paper, we studied the effect of Ptloading on GNS for hydrogen fuel cell. We focus aboutthe ORR catalytic activity of Pt/GNS,it is causedthe ORR at the cathode of fuel cells plays a key role in controlling the performance of a fuel cell, and the efficiencyof ORR electrocatalysts are essential for fuel cells practical applications.21
Experimental
Preparation of grapheneand, Pt/GNS catalysts
Graphene was prepared by the oxidation of graphite powder using the modified Hummers method.22Briefly, graphite powder (0.2 g) (C (98 %), particle size 45 μm, Wako Pure Chemical Industries, Ltd.) and sodium nitrate (NaNO3) (0.16 g) were first stirred in concentrated sulphuric acid (95 wt % H2SO4 ) (6.7 mL) for 2 h while being cooled in an ice water bath. Then, potassium permanganate (KMnO4) (0.9 g) was gradually added to form a new mixture. After 4 h in an ice water bath, the mixture was allowed to stand for 48 h at room temperature with gentle stirring. Thereafter, 20 mL of 5 wt% H2SO4 aqueous solution was added into the above mixture over 1 h with stirring. Then, 0.5 mL of H2O2 (30 wt% aqueous solution) was also added to the above liquid and the mixture was stirred for 2 h. After that, 20 mL of 3 wt% H2SO4/0.5 wt% H2O2 solutions was added into suspension and centrifuged (3000 rpm, 1 h).Subsequently, the product was dispersed in water and ultrasonicated for 5 h. This process affords material of oxidized graphenenanosheets (OGS).23Finally, the OGS were reduced with hydrazine hydrate at room temperature for 48 h. This product was filtered and washed with distilled water and dried in air at RT for 24 h.The as-received powder is the so-called graphenenano sheets (GNS).The detail characterization of GNS can be seen in reference as we reported previously.24
To prepare 10–70wt %Pt/GNScatalysts, the calculated amount of Pt precursor H2PtCl6.6H2O(Alfa Aesar, A Johnson Matthey Company) were dissolvedin 50 mL ethanol. Subsequently, each of ethanol solution of the precursor was mixed with ethanol solution of GNS. After stirring solution for 3 h, each of the products was collected by filtration and dried in air at 60oC for 12 h. Each of them was then reduced by a hydrogenstream 25 mL/min at 400oC for 2h in a furnace. Finally, the catalysts were collected and denoted as 10–70wt %Pt/GNS catalysts. The amount of Pt on GNS was measured by thermogravimetric/differential thermal analysis (TG/DTA) (see in reference 25). This data shows that the Pt atoms of the catalyst precursor are well deposited on GNS.
Then, the 10–70 wt % Pt/GNS were characterized XPS and electrochemical measurement.XPS measurements were carried out using JEOL JPS 9010 TR (X-ray source AlKα, 1486.6 eV; pass energy 50 eV, energy resolution 1.88eV which was calibrated using Ag 3d5/2 by measuring a clean Ag sample, the uncertainty of binding energy ± 0.05 eV). TEM (JEOL JEM-1400 electron microscope was operated at 80 kV, resolution lattice image 0.20 nm, and resolution point image 0.38 nm. TG/DTA measurements were carried out using TG/DTA6300, Seiko Instruments Inc. (Reference: Pt; Air 200 mL/min; T measurement: 50–1000oC; Rate: 10oC/min), respectively.
Electrochemical Measurement
The ORR activities of GNS and 10–70wt %Pt/GNScatalysts, respectively were assessed by using cyclic voltammetry (CV), and rotating ring disk electrode (RRDE) (PGSTAT PG12, AUTOLAB Potensiostat/Galvanostat) measurements in 0.1 M HClO4. The catalyst ink was prepared bydispersing 1 mg catalyst in the mixture of 500 μL(1:50 in methanol) 5 wt% Nafion solutions (Aldrich), then the mixtureof catalyst ink was sonicated for 60 minutes. Then, 10μL of catalyst ink was transferred onto the polishedglassy carbon disk (diameter = 5 mm, geometric area = 0.283 cm2) and dried to form a thin catalyst layer. The catalysts loading on the glassy carbon are 20 μg for GNS.
The Pt loadings of catalysts on the glassy carbon are 2.0, 3.0, 4.0, 5.0, 6.0, 7.0, 8.0, 9.0, 10.0, 11.0, and 12.0μgfor 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, and 70 wt % Pt/GNS, respectively. The CV measurement was carried out by using a typical three-electrode systems consist of a working electrode (glassy carbon), a Pt wire asa counter electrode, and a reversible hydrogen electrode (RHE) as a referenceelectrode. All measurements were performed at room temperature(∼25 oC) using a fresh electrolyte solution (0.1 M HClO4, Sigma–Aldrich). First, the catalyst on the working electrode was purged by bubbling nitrogen (N2) gas at200 mL min−1 through 0.1 M HClO4 for 20 minutes. Then, it was scanned at 0.05–1.0 V versus RHE for 50 cycles with scan rate 10 mVs-1and rotation rate 500 rpm in N2 to eliminate contaminant and de-oxygenates the environment.After that, the saturation gas was switched to oxygen (O2) forRRDE measurement, and the electrolyte was saturated for the samecondition as CV measurement. The RRDE polarization curves were obtained at 0.05–1.0 V versus RHEwith scan rate 10 mV s−1 in O2 saturated 0.1 M HClO4.The commercial catalysts of 20 and 40 wt % Pt/carbon black (CB) (Johnson Matthey) were used as comparison.
Results and discussion
ORR of 10–70 wt % Pt/GNS, and Pt/CB commercial catalysts
Figure 1a and b shows the ORR polarization curves and ORR catalytic activity for 10–70 wt% Pt/GNS and 20 and 40 wt % Pt/CB commercialcatalysts, respectively.In the case of 10, 15 and 20 wt% Pt/GNS, catalysts, the ORR catalytic activity increases with increasing Pt amount on GNS (0.93 to 0.99 V versus RHE). Then, the ORR catalytic activity gradually decreases with increasing Pt amount on GNS for 25–70 wt% Pt/GNS.Their onset potentials are decreased from 0.99 to 0.85 V versus RHE for 25–70 wt% Pt/GNS. It indicates that the ORR activities tend to decrease with increasing Pt particle size (Figure 1b). The current density was measured at 0.9 V versus RHE for 10–70 wt% Pt/GNS and 20 and 40 wt % Pt/CB commercialcatalysts are shown in Figure 2. In the case of 10–20wt% Pt/GNS catalysts, the current densities increase with increasing Pt amount (0.03 to 0.9 V versus RHE). Then, their current densities decrease with increasing Pt amount for 25–70wt % Pt/GNS catalysts. It is also consistent with the ORR data. These data indicate that Pt particle size affects the ORR activity for Pt/GNS catalysts. Interestingly, 20wt % Pt/GNS catalyst shows the higher ORR catalytic activity than 20 wt %Pt/CB commercial catalyst. The 20 wt % Pt/GNS exhibits the highest ORR activity from its onset potential (0.99 V versus RHE) and half wave potential (E1/2 ≈ 0.88 V versus RHE) among the other catalysts. This is also consistent with its current density 0.9 to 0.4 mA cm-2 measured at 0.9 V versus RHE, for 20 wt % Pt/GNS and Pt/CB, respectively. This is a good new where the GNS is better than CB as a supporting material for cathode catalyst.
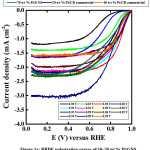 |
Figure 1a: RRDE polarization curves of 10–70 wt % Pt/GNS |
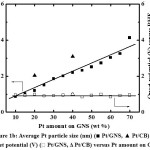 |
Figure 1b: Average Pt particle size (nm) (■ Pt/GNS, ▲ Pt/CB) and onset potential (V) (□ Pt/GNS, Δ Pt/CB) versus Pt amount on GNS. |
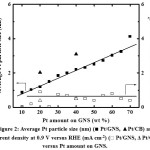 |
Figure 2: Average Pt particle size (nm) (■ Pt/GNS, ▲Pt/CB) and current density at 0.9 V versus RHE (mA cm-2) (□ Pt/GNS, Δ Pt/CB) versus Pt amount on GNS. Click here to View Figure |
XPS
Figure 3, 3aand 4 show therelationship between Pt binding energies and particle size based on XPS measurement for 10–70 wt % Pt/GNS and 20 and 40 wt % Pt/CB commercial catalyst. It shows that the binding energies (BEs) decrease with increasing Pt particle size. In addition, the BE of Pt on Pt/CB is close to the bulk Pt (71.2 eV),26 indicating the weak interaction between Pt and CB. Interestingly, the BEs of Pt on Pt/GNS shifts to higher binding energy compare to Pt/CB. It indicates that the strong interaction between Pt and GNS. Therefore, the GNS can be expected to modify the Pt electronic structure. The relationship between Pt binding energy and ORR catalytic activity for 10–70 wt % Pt/GNS and 20 and 40 wt % Pt/CB commercial catalysts is shown in Figure 4. It clearly shows that the Pt binding energy is linearly related to onset potential. This data also consistent with Pt binding energy versus current density (Figure 5).Thesedataindicate that the strong interaction between Pt and GNS, probably π–d interaction mayaffect the oxygen reduction catalytic activity. The 20 wt % Pt/GNS catalystis very special because it has highest the ORR catalytic activity among the others. It is possible due to in this catalyst condition, the optimum condition for oxygen reduction can be obtained.
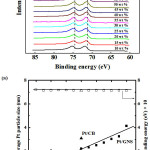 |
Figure 3: XPS spectra of 10–70 wt % Pt/GNS and 20 and 40 wt % Pt/CB commercial catalysts in the Pt 4f region. The results for the 20 and 40 wt % Pt/CB commercial catalysts are also shown. |
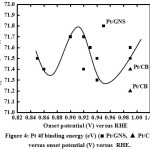 |
Figure 4: Pt 4f binding energy (eV) (■ Pt/GNS, ▲ Pt/CB) versus onset potential (V) versus RHE. |
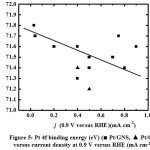 |
Figure 5: Pt 4f binding energy (eV) (■ Pt/GNS, ▲ Pt/CB) versus current density at 0.9 V versus RHE (mA cm-2) |
 |
Figure 6: Current density at 0.9 V versus RHE (mA cm-2) (■ Pt/GNS, ▲ Pt/CB) versus Pt 4f binding energy (eV). |
Conclusions
In this study, we used GNS as a support material to depositedPt particles in order to investigate the support material effect for ORR activity.We found that the ORR activity of Pt/GNS is higher than Pt/CB commercial catalyst. It indicates that the Pt electronic structure is modified by GNS. Therefore, the GNS as supporting material is better than CB for properties of Pt catalysts.
References
- Shao, Y.; Liu, J.; Wang, Y.; Lin, Y. Novel catalyst support materials for PEM fuel cells: current status and future prospects. J. Mat. Chem.2009, 19, 46–59.
CrossRef - Gasteiger, H. A.; Markovic, N. M.Just a Dream – or Future Reality?.Science2009, 324, 48–49.
CrossRef - Gasteiger, H. A.; Kocha, S. S.; Sompalli, B.; Wagner, Frederick T. Activity benchmarks amd requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B. Environ.2005, 56,9–35.
CrossRef - Peng, Z.; Yang, H. Designer platinum nanoparticles: Control of shape, composition in alloy, nanostructure and electrocatalytic property. Nano Today2009, 4, 143–164.
CrossRef - Markovic, N. M.; Schmidt, T. J.; Stamenkovic, V.; Ross, P. N. Fuel Cells2001, 1, 105–116.
CrossRef - Berger,D.J. Science1999, 286, 49–50.
CrossRef - Kou,R.; Shao,Y.; Mei,D.;Nie,Z.; Wang,D.; Wang,C.;Viswanathan,V. V.; Park,S.;Aksay, I. A.; Lin, Y.; Wang,Y.;Liu,J. J. Am. Chem. Soc.2011, 133, 2541–2547.
CrossRef - Shao, Y.; Yin, G.; Gao, Y. J. Power Sources2007, 171, 558–566.
CrossRef - Zhou, J.G.; Zhou,X. T.; Sun,X. H.; Li, R. Y.; Murphy, M.; Ding,Z. F.; Sun, X. L.; Sham, T. K.Chem. Phys. Lett., 2007, 437, 229–232.
CrossRef - Shao, Y.; Sui, J.; Yin, G.; Gao, Y. Appl. Catal., B2008, 79, 89–99.
CrossRef - Yoshitake,T.;Shimakawa,Y.;Kuroshima,S.; Kimura,H.;Ichihashi, T.; Kubo,Y.;Kasuya,D.; Takahashi,K.;Kokai,F.;Yudasaka,M.;Iijima. S. Physica B, 2002, 323, 124–126.
CrossRef - Park,K.W.; Sung,Y.E.; Han,S.; Yun,Y.;Hyeon,T. J. Phys. Chem. B, 2004,108, 939–944
CrossRef - Shao,Y.Y.; Yin,G.P.; Wang,J.J.;Gao,Y.Z.; Shi, P.F. J. Power Sources, 2006,161, 47–53.
CrossRef - Bessel,C.A.;Laubernds,K.;Rodriguez,N.M.;Baker R.T.K. J. Phys. Chem. B, 2001,105, 1115–1118.
CrossRef - Wang, J.; Yin, G.; Shao, Y.; Zhang, S.; Wang, Z.; Gao, Y. J. Power Sources2007, 171331–339.
- Sun,C.L.; Chen,L.C.; Su,M.C.; Hong,L.S.;Chyan, O.; Hsu,C.Y.; Chen,K.H.; Chang, T.F.; Chang, L. Chem. Mater., 2005, 17, 3749–3753.
CrossRef - Shao, Y.Y.;Yin, G.P.; Wang,Z.B.;Gao,Y.Z. J. Power Sources, 2007, 167, 235–242.
CrossRef - Stoller, M. D.; Park, S.; Zhu, Y.; An, J.; Ruoff, R. S. Nano Lett.2008, 8, 3498–3502.
CrossRef - Novoselov, K. S.; Geim, A.; Morozov, S. V.; Jiang, D.; Zhang, Y.; Dubonos, S. V.; Grigorieva, I. V.; Firsov, A. A. Science2004, 306, 666–669.
CrossRef - Antolini, E. Applied Catalysis B: Environmental2012,123–124, 52–68.
CrossRef - Cherstiouk, O. V.; Simonov, P. A.; Savinova, E. R. Electrochim.Acta2003, 48, 3851–3860.
CrossRef - Hummers, W. S.; Offeman, R. E. J. Am. Chem. Soc., 1958, 80, 1339.
CrossRef - Stankovich, S.; Dikin, D. A.; Dommett, G. H. B.; Kohlhaas, K. M.; Zimney, E. J.; Stach, E. A.; Piner, R. D.; Nguyen, S. T. ;Ruoff, R. S. Nature2006, 442, 282–286.
CrossRef - Siburian, R.; Nakamura, J. J. Phys. Chem. C 2012, 116, 22947–22953.
CrossRef - Joo, J. B.; Kim, P.; Kim, W.; Kim, Y.; Yi, J. J Appl. Electrochem.2009, 39, 135–140.
CrossRef - Vidakovic, T.; Christov, M.; Sundmacher, K. Electrochim. Acta2007, 52, 5606–5613.
CrossRef - Cherstiouk, O. V.; Simonov, P. A.; Savinova, E. R. Electrochim.Acta2003, 48, 3851–3860.
CrossRef - Moulder, J.; Stickle, W. F.; Sobol, P. E.; Bomben, K. D. Handbook of X-ray Photoelectron Spectroscopy; Perkin Elmer Corporation: Minnesota, USA, 1992; pp. 6, 8, 40–43, 78.
- Qu, L.; Liu, Y.; Baek, J. B.; Dai, L. ACS Nano2010, 4, 1321–1326.
CrossRef - Imamura, G.; Saiki, K. J. Phys. Chem. C.2011, 115, 10000–10005.
CrossRef - Gong, K.; Du, F.; Xia, Z.; Durstock, M.; Dai, L. Science2009, 323,760–764.
CrossRef - He, D.; Cheng, K.; Li, H.; Peng, T.; Xu, F.; Mu, S.; Pan, M. Langmuir2012, 28, 3979–3986.
CrossRef - Shao, M.; Peles, A.; Shoemaker, K. Nano Lett.2011, 11, 3714–3719.
CrossRef - Pozio, A.; De Francesco, M.; Cemmi, A.; Cardellini, F.; Giorgi, L. J. Power Sources2002, 105, 13–19.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


