Design and Synthesis of Two Steroid-Pyrrolo-Triazecin Derivatives Using Estrone and Pregnenolone as Chemical Tools
Lauro Figueroa-Valverde1*, Marcela Rosas-Nexticapa2, catalina cervantes-ortega2, Francisco Díaz-Cedillo3, Elodia García-Cervera1 and Eduardo Pool-Gómez1
1Laboratory of Pharmacochemistry, University Autonomous of Campeche.Av. Agustín Melgar, Col Buenavista C.P. 24039, Campeche Cam., Mexico.
2Universidad Veracruzana. Médicos y Odontólogos s/n, 91010, Xalapa, Veracruz, México.
3Escuela Nacional de Ciencias Biológicas del Instituto Politécnico Nacional. Prol. Carpio y Plan de Ayala s/n Col. Santo Tomas, México, D.F. C.P. 11340.
Corresponding Author E-mail: lauro_1999@yahoo.com
DOI : http://dx.doi.org/10.13005/ojc/330106
Several steroid-pyrrole derivatives have been prepared using different protocols; nevertheless, have some drawbacks such as the use of some reagents that have limited stability and require special conditions. The aim of study involved the synthesis of two steroid-pyrrolo-triazecin derivatives using a series of reactions that involve; cycloaddition [2 + 2], displacement of nitro group, formation of shift base, amidation/cyclization using boric acid (Method A) or carbodiimidederivative (Method B). The results showed that there is a higher yielding with the method A in comparison with the method B. In addition, chemical structure of compounds was confirmed by NMR spectroscopic data.
KEYWORDS:Estrone; pregnenolone; pyrrol; triazecin; steroid
Download this article as:| Copy the following to cite this article: Figueroa-Valverde L, Rosas-Nexticapa M, cervantes-ortega C, Díaz-Cedillo F, García-Cervera E, Pool-Gómez E. Design and Synthesis of Two Steroid-Pyrrolo-Triazecin Derivatives Using Estrone and Pregnenolone as Chemical Tools. Orient J Chem 2017;33(1). |
| Copy the following to cite this URL: Figueroa-Valverde L, Rosas-Nexticapa M, cervantes-ortega C, Díaz-Cedillo F, García-Cervera E, Pool-Gómez E. Design and Synthesis of Two Steroid-Pyrrolo-Triazecin Derivatives Using Estrone and Pregnenolone as Chemical Tools. Orient J Chem 2017;33(1). Available from: http://www.orientjchem.org/?p=27846 |
Introduction
For several years there has been interest on the preparation of some substituted pyrroles; for example, the synthesis of the reaction of 1,4-diols with amines to form substituted pyrroles was developed1. Other data2 showed the synthesis of aryl-substituted pyrroles using the three-component system (arylketones, 1,2-diols and amines) in presence of ruthenium. In addition, a pyrrole-steroid derivative3 was prepared by reaction of androsterone-arylidine with acenaphthylene-1,2-dione and 1,3-thiazolane-4-carboxylic acid. Other study showed a reaction regioselective palladium-catalyzed cyclization of internal alkynes and 2-amino-3-iodoacrylates to form some substituted pyrroles4. Also, another report indicated that the cationic N-heterocyclic carbine-gold(I) complex catalyzes the formation of tri- and tetrasubstitutedpyrroles via the amino-Claisen rearrangement of N-propargyl β-enaminone derivatives and the cyclization of α-allenyl β-enaminone as intermediates5. Additionally, a study showed that a Lewis acid can activate to cyclopropanes which react with aliphatic, aromatic, and α,β-unsaturated nitriles via dipolar cycloaddition [3 + 2], following by dehydration, and a tautomerization sequence for preparation of pyrroles6. Another report indicated that the Dirhodium(II) reagent can used in the three-component system (imine, diazoacetonitrile, and an activated alkynyl) as catalyst to form substituted 1,2-diarylpyrroles7.In addition, a study showed that Au(I) reagent Catalyzed the reaction of aryl-substituted with N-tosylalkynylaziridines to give 2,5-substituted pyrroles8. Another study showed a rearrangement reaction of a propargylic-aziridine, catalyzed by PPh3AuCl/AgOTf, forming some pyrrole derivatives9. Additionally, a report showed a direct synthesis of pyrroles by the reaction of imines, acid chlorides, and alkynes catalyzed by isocyanides is reported10. Other data showed the functional homologation of a β-ketoester with an aldehyde followed by an oxidation to form a series of different substituted 1,4-dicarbonyl compounds that can be rapidly cyclized with the Paal-Knorr procedure for synthesis some pyrrole derivatives11. All these experimental results show different methods for the preparation of unsubstitutedpyrrole; nevertheless. It is important tomention that there aresomeprotocolsthat requirehazardous reagentsas well asdifferent experimental conditionsfor the preparationof these compounds. Therefore, in this study two steroid-pyrrolo-triazecin derivatives were prepared using some chemical tools.
Material and Methods
General Chemical
The compounds evaluated in this study were purchased from Sigma-Aldrich Co., Ltd. The melting point of compounds was determined on an Electrothermal (900 model). Infrared spectra (IR) were recorded using KBr pellets on a Perkin Elmer Lambda 40 spectrometer. 1H and 13C NMR (nuclear magnetic resonance) spectra were recorded on a Varian VXR-300/5 FT NMR spectrometer at 300 and 75.4 MHz (megahertz) in CDCl3 (deuteratedchloform) using TMS (tetramethylsilane) as internal standard. EIMS (electron impact mass spectroscopy) spectra were obtained with a Finnigan Trace Gas Chromatography Polaris Q Spectrometer. Elementary analysis data were acquired from a Perkin Elmer Ser. II CHNS/0 2400 elemental analyzer.
Synthesis of 4-(4-nitrophenyl)1,3.dihydro-2H-pyrrol-2-one (5)
A solution of 4-nitrophenylacetonitrile (200 mg, 1.23 mmol), 6-cloro-1-hexyne (180 µl, 1.38 mmol), and triethylamine (220 µL, 1.56 mmol) in 5 mL of chloroform was stirred for 72 h at room temperature. The reaction mixture was evaporated to dryness under reduced pressure, the residue washed 3 times with water. Then the precipitate was separated and dried at room temperature. The product yield was 66 %; m.p. 44-46 oC; 1670 and 1600; 1H NMR (300 MHz, CDCl3) dH: 3.22 (m, 2H), 7.28 (d, 1H), 7.44 (m, 2H), 8.02 ( broad, 1H), 8.10 (m, 2H) ppm. 13C NMR (75.4 Hz, CDCl3) dC: 42.20 (C-3), 117.32 (C-4), 121.54 (C-9, C-11), 124.63 (C-5), 132.54 (C-8, C-12), 144.30 (C-10), 144.76 (C-7), 176.98 (C-2) ppm. EI-MS m/z: 204.05. Anal. Calcd. for C10H8N2O3: C, 58.82; H, 3.95; N, 13.72; O, 23.51. Found: C, 58.74; H, 3.82.
Preparation of steroid-oxy-phenyl-acetonitrile derivatives
A solution of 1 or 2 (0.5 mmol), 4-nitrophenylacetonitrile (90 mg, 0.55 mmol), potassium carbonate (40 mg, 0.30 mmol) in 5 mL of dimethyl sulfoxide was stirred for 72 h at room temperature. The reaction mixture was evaporated to dryness under reduced pressure, the residue washed 3 times with water. Then the precipitate was separated and dried at room temperature.
2-(4-(((13S)-13-methyl-17-oxo-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a] phenanthren-3-yl)oxy)phenyl)acetonitrile (6)
The product yield was 52 %; m.p. 202-204 oC; 2269, 1710 and 1246; 1H NMR (300 MHz, CDCl3) dH: 1.00 (s, 3H), 1.22-1.36 (m, 4H), 1.54-1.94 (m, 3H), 2.08-2.20 (m, 4H), 2.48-3.00 (m, 4H), 3.66 (m, 2H), 6.70-6.78 (m, 2H), 6.98 (m, 2H), 7.14 (m, 1H), 7.36 (m, 2H). 13C NMR (75.4 Hz, CDCl3) dC: 13.60 (C-18), 21.78 (C-7), 23.50 (C-27), 25.72 (C-3), 26.32 (C-10), 29.40 (C-11), 30.92 (C-4), 35.70 (C-8), 37.56 (C-1), 44.70 (C-2), 48.00 (C-5), 48.62 (C-6), 114.26 (C-17), 114.51 (C-15), 117.10 (C-22, C-26), 117.40 (C-28), 122.50 (C-24), 124.60 (C-14), 128.30 (C-23, C-25), 132.61 (C-13), 139.54 (C-12), 155.00 (C-21), 155.94 (C-16), 219.80 (C-9) ppm. EI-MS m/z: 385.20. Anal.Calcd.for C26H27NO2: C, 81.01; H, 7.06; N, 3.63; O, 8.30. Found: C, 81.00; H, 7.00.
[4-(17-acetyl-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H cyclopenta [a]phenanthren-3-yl)oxy)phenyl]acetonitrile (7)
The product yield was 52 %; m.p. 168-170 oC; 2258, 1712 and 1248; 1H NMR (300 MHz, CDCl3) dH: 0.64 (s, 3H), 1.00 (s, 3H), 1.04-1.38 (m, 5H), 1.44-2.08 (m, 11H), 2.10 (s, 3H), 2.20-2.54 (m, 4H), 3.66 (t, 2H, J = 18.24 Hz), 4.20 (m, 1H), 5.46 (d, 1H, J = 2.00 Hz), 6.84-7.28 (m, 4H). 13C NMR (75.4 Hz, CDCl3) dC: 13.23 (C-25), 23.42 (C-28), 24.00 (C-15), 25.66 (C-3), 26.15 (C-10), 30.22 (C-31), 31.40 (C-14), 32.56 (C-2), 32.66 (C-16), 36.20 (C-6), 36.88 (C-9), 41.58 (C-11), 43.76 (C-5), 44.60 (C-8), 47.43 (C-4), 54.50 (C-7), 63.70 (C-17), 80.32 (C-1), 115.20 (C-20, C-24), 117.40 (C-29), 122.24 (C-13), 122.80 (C-22), 127.10 (C-21, C-23), 139.00 (C-12), 156.32 (C-19), 209.23 (C-26) ppm. EI-MS m/z: 417.26. Anal.Calcd.for C30H37NO3: C, 80.53; H, 8.45; N, 3.35; O, 7.86. Found: C, 80.44; H, 8.36.
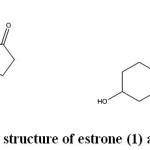 |
Figure 1: Chemical structure of estrone (1) and pregnenolone (2). |
Preparation of steroid-phenyl-pyrrolone derivatives
Method A
A solution of 6 or 7 (0.5 mmol), 6-cloro-1-hexyne (94 ml, 0.77 mmol), potassium carbonate anhydride (50 mg, 0.36) in 5 mL of dimethyl sulfoxide was stirred for 72 h at room temperature. The reaction mixture was evaporated to dryness under reduced pressure, the residue washed 3 times with water. Then the precipitate was separated and dried at room temperature.
4-(4-(((13S)-13-methyl-17-oxo-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a] phenanthren-3-yl)oxy)phenyl)1,5-dihydro-2H-pyrrol-2-one (8).
The product yield was 45 %; m.p. 202-204 oC; 1705, 1672 and 1600; 1H NMR (300 MHz, CDCl3) dH: 1.00 (s, 3H), 1.22-1.36 (m, 4H), 1.54-1.94 (m, 3H), 2.08-2.20 (m, 4H), 2.48-3.00 (m, 4H), 4.56 (m, 2H), 5.50 (d, 1H, J = 2.10 Hz), 6.70 (m, 1H), 6.74 (m, 2H), 6.78-7.14 (m, 2H), 7.28 (m, 2H), 8.01 (broad, 1H). 13C NMR (75.4 Hz, CDCl3) dC: 13.60 (C-31), 21.78 (C-25), 25.72 (C-21), 26.32 (C-18), 29.40 (C-17), 30.92 (C-22), 35.70 (C-26), 37.56 (C-19), 44.70 (C-20), 48.00 (C-23), 48.62 (C-24), 50.70 (C-5), 114.26 (C-15), 114.51 (C-28), 117.58 (C-9, C-1), 118.00 (C-3), 124.60 (C-29), 128.60 (C-8, C-12), 130.00 (C-7), 132.61 (C-30), 139.54 (C-16), 154.82 (C-14), 156.91 (C-10), 168.60 (C-4), 174.38 (C-2), 219.80 (C-27) ppm. EI-MS m/z: 427.21. Anal.Calcd.for C28H29NO3: C, 78.66; H, 6.84; N, 3.28; O, 11.23. Found: C, 78.54; H, 6.76.
4-[4-(17-acethyl-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a] phenanthren-3-yl)oxy)phenyl)1,5-dihydro-2H-pyrrol-2-one (9)
The product yield was 52 %; m.p. 168-170 oC; 1702, 1670 and 1246; 1H NMR (300 MHz, CDCl3) dH: 0.64 (s, 3H), 1.00 (s, 3H), 1.04-1.46 (m, 7H), 1.60-2.08 (m, 9H), 2.10 (s, 3H), 2.20-4.20 (m, 5H), 4.56 (m, 2H), 5.44 (d, 1H, J = 2.00 Hz), 5.50 (d, 1H, J = 2.10 Hz), 6.84-7.14 (m, 4H), 8.02 (broad, 1H). 13C NMR (75.4 Hz, CDCl3) dC: 13.23 (C-32), 19.40 (C-31), 21.40 (C-23), 23.10 (C-35), 23.98 (C-28), 30.30 (C-15), 31.44 (C-29), 31.80 (C-19, C-27), 37.56 (C-16), 38.10 (C-17), 38.90 (C-22), 40.00 (C-24), 44.76 (C-21), 50.00 (C-18), 50.66 (C-5), 56.84 (C-20), 63.70 (C-30), 81.42 (C-14), 114.94 (C-9, C-11), 118.00 (C-3), 122.04 (C-26), 127.10 (C-8, C-12), 128.10 (C-7), 140.00 (C-25), 156.44 (C-10), 168.62 (C-4), 174.44 (C-2), 209.23 (C-32) ppm. EI-MS m/z: 473.29. Anal.Calcd.for C31H39NO3: C, 78.61; H, 8.30; N, 2.96; O, 10.13. Found: C, 78.54; H, 8.24.
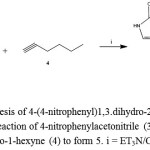 |
Figure 2: Synthesis of 4-(4-nitrophenyl)1,3.dihydro-2H-pyrrol-2-one (5). Reaction of 4-nitrophenylacetonitrile (3) with6-cloro-1-hexyne (4) to form 5. i = ET3N/CHCl3 |
Method B
A solution of 1 or 2 (0.5 mmol), 5 (110 mg, 0.54 mmol), potassium carbonate anhydride (50 mg, 0.36) in 5 mL of dimethyl sulfoxide was stirred for 72 h at room temperature. The reaction mixture was evaporated to dryness under reduced pressure, the residue washed 3 times with water. Then the precipitate was separated and dried at room temperature. The yield for 8 was 65 % and for 9 of72%. Similar 1H NMR and 13C NMR data for the compound 8 or 9 were obtained compared with method A.
Preparation of pyrrol-ethanediamine derivatives
A solution of 8 or 9 (0.5 mmol), ethylenediamine(60 µl, 0.90 mmol), boric acid (50 mg, 0.80 mmol) in 5 mL of methanol was stirred for 72 h at room temperature. The reaction mixture was evaporated to dryness under reduced pressure, the residue washed 3 times with water. Then the precipitate was separated and dried at room temperature.
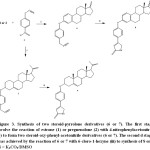 |
Figure 3: Synthesis of two steroid-pyrrolone derivatives (6 or 7).The first stage involve the reaction of estrone (1) or pregnenolone (2) with 4-nitrophenylacetonitrile (3) to form two steroid-oxy-phenyl-acetonitrile derivatives (6 or 7). The second d stage was achieved by the reaction of 6 or 7 with 6-cloro-1-hexyne (iii) to synthesis of 8 or 9. ii = K2CO3/DMSO |
N1-(4-{4-[17-(2-amino-ethylimino)-13-methyl-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-3-yloxy]-phenyl}-1,5-dihydro-pyrrol-2-ylidene)-ethane-1,2-diamine (11)
The product yield was 52 %; m.p. 124-126 oC; 3380, 3320 and 1248; 1H NMR (300 MHz, CDCl3) dH: 1.00 (s, 3H), 1.22-1.90 (m, 6H), 2.08-2.80 (m, 9H), 3.10 (t, 2H, J = 13.34 Hz), 3.16 (t, 2H, J = 13.34 Hz), 3.50 (t, 2H, J = 8.79 Hz), 3.66 (t, 2H, J =8.79 Hz), 4.90 (m, 2H) 5.30 (broad, 5H), 6.68 (m, 1H), 6.70 (d, 1H, J = 2.10 Hz), 6.74 (m, 2H), 6.78 (m, 1H), 7.24 (m, 2H), 7.30 (m, 1H). 13C NMR (75.4 Hz, CDCl3) dC: 15.80 (C-34), 22.00 (C-28), 25.82 (C-21), 26.00 (C-24), 27.50 (C-29), 29.76 (C-20), 32.42 (C-25), 37.56 (C-22), 40.94 (C-8), 41.00 (C-37), 41.44 (C-26), 43.32 (C-23), 54.00 (C-7), 54.14 (C-36), 54.30 (C-27), 59.52 (C-5), 114.26 (C-18), 114.51 (C-31), 118.98 (C-12, C-14), 124.20 (C-3), 125.88 (C-32), 129.10 (C-11, C-15), 130.61 (C-10), 134.14 (C-33), 140.80 (C-19), 153.32 (C-4), 154.78 (C-2), 154.80 (C-17), 158.14 (C-13), 176.80 (C-30) ppm. EI-MS m/z: 511.33. Anal. Calcd. for C32H41N5O: C, 75.11; H, 8.08; N, 13.69; O, 3.13. Found: C, 75.04; H, 8.00.
N1-[4-(4-{17-[1-(2-amino-ethylimino)-ethyl]-13-methyl-2,3,4,7,8,9,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yloxy}-phenyl)-1,5-dihydro-pyrrol-2-ylidene]-ethane-1,2-diamine (12)
Theproductyieldwas 52 %; m.p. 120-122 oC; 3380, 3318 and 1246; 1H NMR (300 MHz, CDCl3) dH: 0.90 (s, 3H), 0.98 (m, 1H), 1.22-1.70 (m, 9H), 1.80 (s, 3H), 1.82-2.50 (m,10), 3.10 (t, 2H, J = 13.34 Hz), 3.16 (t, 2H, J = 13.34 Hz), 3.50 (t, 2H, J = 8.79 Hz), 3.66 (t, 2H, J = 8.79 Hz), 4.20 (m, 1H) 4.18 (m,1), 4.90 (m, 2H), 5.28 (broad, 5H), 5.44 (d, 1H, J = Hz), 6.70 (d, 1H, J = 2.10 Hz), 6.78 (m, 2H), 7.14 (m, 2H). 13C NMR (75.4 Hz, CDCl3) dC: 13.30 (C-34), 16.70 (C-40), 19.40 (C-34), 21.22 (C-26), 26.40 (C-32), 26.60 (C-31), 30.30 (C-18), 30.78 (C-22), 31.90 (C-30), 37.56 (C-19), 38.10 (C-20), 38.18 (C-25), 40.00 (C-27), 40.94 (C-8), 41.00 (C-39), 42.82 (C-24), 50.40 (C-21), 53.10 (C-38), 54.02 (C-7), 55.30 (C-23), 59.52 (C-5), 63.10 (C-33), 81.40 (C-17), 116.26 (C-12, C-14), 122.11 (C-29), 124.20 (C-3), 127.58 (C-11, C-15), 128.80 (C-10), 140.02 (C-28), 153.32 (C-4), 154.78 (C-2), 156.70 (C-35), 157.74 (C-13) ppm. EI-MS m/z: 557.40. Anal. Calcd. for C35H51N5O: C, 75.36; H, 9.22; N, 12.56; O, 2.87. Found: C, 75.22; H, 9.12.
Preparation of carboxy-propionylamino-steroid- succinamic acid
A solution of 11 or 12 (0.50 mmol), ethylenediamine (144 mg, 1.22 mmol) and N-(3- dimethylaminopropyl)-N’-ethylcarbodiimide (90 mg, 0.58 mmol) in 10 mL of methanol was stirred for 72 h at room temperature. The reaction mixture was evaporated to a smaller volume. Then the mixture was diluted with water and extracted with chloroform. The organic phase was evaporated to dryness under reduced pressure, the residue was purified by crystallization from methanol:water (3:1)
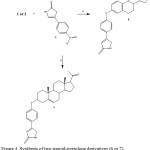 |
Figure 4: Synthesis of two steroid-pyrrolone derivatives (6 or 7).Reaction of estrone (1) or pregnenolone (2) with 5 to form 6 or 7.iv = K2CO3/DMSO |
N-{2-[3-(4-{5-[2-(3-carboxy-propionylamino)-ethylimino]pyrrolidin-3-yl}-phenoxy)-13-methyl-6,7,8,9,11,12,13,14,15,16-decahydro-cyclopenta[a]phenanthren-17-ylideneamino]-ethyl}-succinamic acid (14)
The product yield was 52 %; m.p. 164-166 oC; 3320, 1674 and 1248; 1H NMR (300 MHz, CDCl3) dH: 1.00 (s, 3H), 1.22-1.90 (m, 6H), 2.06-2.28 (m, 5H), 2.50 (t, 2H, J = 13.34 Hz), 251 (t, 2H, J = 15.34 Hz), 2.54 (t, 2H, J = 16.80 Hz), 2.55 (t, 2H, J = 16.80 Hz), 2.60-2.80 (m, 4H), 3.32 (t, 2H, J = 15.00 Hz), 3.54 (t, 2H, J = 15.00 Hz), 3.55 (t, 2H, J = 15.00 Hz), 3.70 (t, 2H, J =8.79 Hz), 4.90 (m, 2H), 6.68 (m, 1H), 6.70 (d, 1H, J = 2.10 Hz), 6.72 (m, 2H), 6.78 (m, 1H), 7.24 (m, 2H), 7.30 (m, 1H), 8.40 (broad, 5H) ppm. 13C NMR (75.4 Hz, CDCl3) dC: 15.80 (C-41), 22.00 (C-34), 25.82 (C-27), 26.00 (C-30), 27.50 (C-35), 28.84 (C-18, C-48), 29.76 (C-26), 30.00 (C-19, C-49), 32.42 (C-31), 36.00 (C-19), 36.08 (C-44), 37.56 (C-28), 41.44 (C-32), 43.32 (C-29), 54.10 (C-7), 54.24 (C-43), 54.30 (C-33), 59.52 (C-5), 114.26 (C-24), 114.51 (C-37), 118.98 (C-14, C-16), 124.20 (C-3), 125.88 (C-38), 129.10 (C-13, C-17), 130.61 (C-12), 134.14 (C-39), 140.80 (C-25), 153.32 (C-4), 154.78 (C-2), 154.80 (C-23), 158.14 (C-15), 166.80 (C-10, C-46), 173.56 (C-20, C-50), 176.88 (C-50) ppm. EI-MS m/z: 711.36. Anal. Calcd. for C40H49N5O7: C, 67.49; H, 6.94; N, 9.84; O, 15.73. Found: C, 67.37; H, 6.90.
N-(2-{4-[4-(17-{1-[2-(3-carboxy-propionylamino)-ethylimino]-ethyl}-13-methyl-2,3,4, 5,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yloxy)-phenyl]1,5-dihydro-pyrrol-2-ylideneamino}-ethyl)-succinamic acid (15)
Theproductyieldwas 52 %; m.p. 178-180; 3318, 1700, 1674 and 1248; 1H NMR (300 MHz, CDCl3) dH: 0.78, (s, 3H), 0.90 (s, 3H), 1.30-1.56 (m, 5H), 1.68 (s, 3H), 1.70-1.94 (m, 6H), 2.00-2.42 (m, 8H), 2.50 (t, 2H, J = 13.34 Hz), 2.51 (t, 2H, J = 13.34 Hz), 2.55 (t, 2H, J = 8.79 Hz), 2.56 (t, 2H, J = 8.79 Hz), 3.46 (t, 2H, J = 13.34 Hz), 3.54 (t, 2H, J = 13.34 Hz), 3.60 (t, 2H, J = 13.34 Hz), 3.74 (t, 2H, J = 13.34 Hz), 4.00 (m, 1H), 4.88 (m, 2H), 5.40-5.60 (m, 2H), 6.70 (d, 1H, J = 2.10 Hz), 6.80 (m, 2H), 7.12 (m, 2H), 8.14 (broad 5H) ppm. 13C NMR (75.4 Hz, CDCl3) dC: 14.64 (C-41), 16.36 (C-42), 21.22 (C-32), 22.58 (C-50), 26.20 (C-38), 28.90 (C-18, C-24, C-51), 30.00 (C-19, C-52), 34.08 (C-33), 34.56 (C-26), 35.00 (C-25), 35.70 (C-46), 36.00 (C-8), 37.46 (C-28), 37.96 (C-31), 43.56 (C-30), 45.54 (C-34), 51.08 (C-27), 53.00 (C-29), 53.30 (C-45), 54.10 (C-7), 57.00 (C-39), 59.52 (C-5), 82.66 (C-23), 116.00 (C-14, C-16), 124.21 (C-3), 127.58 (C-13, C-17), 128.80 (C-12), 129.48 (C-35), 130.80 (C-36), 153.32 (C-4), 154.78 (C-2), 157.40 (C-15), 162.70 (C-43), 166.80 (C-10, C-48), 173.64 (C-20, C-53) ppm. EI-MS m/z: 757.44. Anal. Calcd. for C42H57N5O7: C, 68.14; H, 7.85; N, 9.24; O, 14.78. Found: C, 68.06; H, 7.78.
Preparation of Pyrrolo-steroid-triazecin-oxobutanoic acids complex
Method A
A solution of 14 or 15 (0.50 mmol), N-(3- dimethylaminopropyl)-N’-ethylcarbodiimide (90 mg, 0.58 mmol) in 10 mL of methanol was stirred for 72 h at room temperature. The reaction mixture was evaporated to a smaller volume. Then the mixture was diluted with water and extracted with chloroform. The organic phase was evaporated to dryness under reduced pressure, the residue was purified by crystallization from methanol:water (3:1).
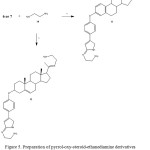 |
Figure 5: Preparation of pyrrol-oxy-steroid-ethanediamine derivatives (11 or 12).Reaction of 6 or 7with ethylenediamineto form 11 or 12.v = boric acid/rt. |
4-((2-(((13S,E)-3-(4-((Z)-5,8-dioxo-2,3,4,5,6,7,8,10,11,12-decahydropyrrolo [1,2-a] [1,3, 6]triazecin-11-yl)-phenoxy)-13-methyl-6,7,8,9,11,12,13,14,15,16-decahydro-17H-cyclo penta[a]phenanthren-17-ylidene)amino)ethyl)amino)-4-oxobutanoic acid (16)
The product yield was 40 %; m.p. 116-118 oC; 3320, 1702, 1672 and 1246; 1H NMR (300 MHz, CDCl3) dH: 1.02 (s, 3H), 1.22-2.18 (m, 9H), 2.22 (t, 2H, J = 13.34 Hz),2.25-2.28 (m, 2H), 2.40 (t, 2H, J = 13.34 Hz), 2.52 (t, 2H, J = 15.34 Hz), 2.56 (t, 2H, J = 16.80 Hz), 2.62-2.80 (m, 4H), 3.32 (t, 2H, J = 15.00 Hz), 3.52 (t, 2H, J = 15.00 Hz), 3.55 (t, 2H, J = 15.00 Hz), 3.88 (t, 2H, J =8.79 Hz), 4.88-5.16 (m, 2H), 6.68 (m, 1H), 6.75 (m, 2H), 6.76 (m, 1H), 7.24 (m, 1H, J = Hz), 7.28 (m, 1H), 7.29 (m, 2H), 7.76 (broad, 1H, -NHCO), 8.58 (broad, 5H, -NHCO and -COOH) ppm. 13C NMR (75.4 Hz, CDCl3) dC: 15.80 (C-40), 22.00 (C-34), 25.82 (C-27), 26.00 (C-30), 27.50 (C-35), 28.84 (C-47), 29.76 (C-26), 30.00 (C-48), 31.60 (C-7), 32.42 (C-31), 33.50 (C-6), 36.08 (C-43), 37.56 (C-28), 37.79 (C-3), 41.44 (C-32), 43.32 (C-29), 54.24 (C-42), 54.30 (C-33), 58.92 (C-13), 62.12 (C-2), 114.26 (C-24), 114.51 (C-37), 119.20 (C-18, C-20), 124.90 (C-11), 125.88 (C-38), 129.10 (C-17, C-21), 130.00 (C-16), 134.14 (C-39), 140.80 (C-25), 147.50 (C-10), 150.66 (C-12), 154.78 (C-23), 158.14 (C-19), 166.80 (C-45), 170.60 (C-5), 173.56 (C-49), 176.88 (C-36), 178.40 (C-8) ppm. EI-MS m/z: 693.35. Anal. Calcd. for C40H47N5O6: C, 69.24; H, 6.83; N, 10.09; O, 13.84. Found: C, 69.12; H, 6.71.
4-((2-(((Z)-1-((10S,13S)-3-(4-((Z)-5,8-dioxo-2,3,4,5,6,7,8,10-decahydropyrrolo[1,2-a][1,3,6]triazecin-11-yl)-phenoxy)-10,13-dimethyl-2,3,4,5,8,,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethylidene)amino)ethyl)amino-4-oxobutanoic acid (17)
Theproductyieldwas 38 %; m.p. 164-166 oC; 3322, 1700, 1676 and 1248; 1H NMR (300 MHz, CDCl3) dH: 0.78 (s, 3H), 1.00 (s, 3H), 1.28-1.56 (m, 5H), 1.68 (s, 3H), 1.70 -2.14 (m, 11H), 2.20 (t, 2H, J = 13.34 Hz), 2.30-2.40 (m, 2H), 2.42 (t, 2H, J = 13.34 Hz), 2.43 (m, 1H), 2.50 (t, 2H, J = 13.34 Hz), 2.56 (t, 2H, J = 13.34 Hz), 3.08-3.30 (m, 2H), 3.48 (t, 2H, J = 13.34 Hz), 3.58 (t, 2H, J = 8.79 Hz), 3.62 (t, 2H, J = 13.34 Hz), 3.80 (m, 1H), 3.96 (t, 2H, J =8.79 Hz), 3.98 (m, 1H), 4.00 (m, 1H), 4.22 (m, 1H), 5.36-5.58 (m, 2H), 6.80-7.00 (m, 3H), 7.74 (broad, 1H, -NHCO), 7.90 (broad, 2H, -NHCO and -COOH) ppm. 13C NMR (75.4 Hz, CDCl3) dC: 14.60 (C-40), 16.30 (C-41), 21.22 (C-32), 22.60 (C-49), 26.20 (C-38), 26.25 (C-37), 28.88 (C-24), 28.90 (C-50), 30.00 (C-51), 31.60 (C-7), 33.50 (C-6), 34.08 (C-33), 34.56 (C-26), 35.00 (C-25), 35.70 (C-45), 37.40 (C-28), 37.94 (C-31), 37.98 (C-3), 43.36 (C-11), 43.40 (C-12), 43.60 (C-30), 45.58 (C-34), 51.00 (C-27), 51.70 (C-10), 53.00 (C-29), 53.32 (C-44), 57.00 (C-39), 62.16 (C-2), 82.70 (C-23), 114.54 (C-18, C-20), 125.90 (C-38), 127.50 (C-16), 128.74 (C-17, C-21), 129.44 (C-35), 130.80 (C-36), 156.08 (C-19), 156.30 (C-13), 162.66 (C-42), 166.80 (C-47), 170.60 (C-5), 173.64 (C-52), 180.98 (C-8) ppm. EI-MS m/z: 741.44. Anal. Calcd. for C43H59N5O6: C, 69.61; H, 8.02; N, 9.44; O, 12.94. Found: C, 69.52; H, 8.00.
Method B
A solution of 14 or 15 (0.50 mmol), boric acid (50 mg, 0.80 mmol) in 10 mL of methanol was stirred for 72 h at room temperature. The reaction mixture was evaporated to a smaller volume. Then the mixture was diluted with water and extracted with chloroform. The organic phase was evaporated to dryness under reduced pressure, the residue was purified by crystallization from methanol:water (3:1), The yield for 8 was 36 % and for 9 of32%. Similar 1H NMR and 13C NMR data for the compounds 16 or 17 were obtained compared with method A.
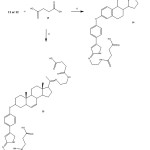 |
Figure 6: Preparation of twocarboxyl-propionylamino-steroid- succinamic acids (14 or 15). Reaction of 11 or 12with succinic acid usinga carbodiimidederivative as catalyst (vi). |
Results and Discussion
There are several methods for preparation of pyrroles; however, despite its wide scope, have some drawbacks such as the use of some reagents that have limited stability and require special conditions; for example, basic medium12, Ru3(CO)1213, ammonia/HCl14and others. In this work two pyrrole derivatives were prepared using several strategies.
Synthesis of 4-(4-nitrophenyl)1,3.dihydro-2H-pyrrol-2-one (5)
The first stage was achieved by the reaction of 4-nitrophenylacetonitrile with 6-cloro-1-hexyne to form a pyrrolone derivative (5) in presence of triethylamine. The 1H NMR spectrum of 5 showed several signals at 3.22-7.28, and 8.02 ppm for pyrrole ring; at 7.44 and 8.10 ppm for phenyl group. The 13C NMR spectrum exhibits the following signals at 42.20-117.32 and 124.63 ppm for pyrrole ring; at 121.54 and 132.54-144.76 ppm for phenyl group; at 176.98 ppm for ketone group. In addition, the mass spectrum of 5 exhibited molecular ion peak atm/z 204.05.
Preparation of steroid-oxy-phenyl-acetonitrile derivatives
The second stage was achieved by preparation of two steroid-3-oxy-phenylacetonitrile derivatives via displacement of nitro group of nitrophenylacetonitrile (3) to form two steroid-oxy-phenyl-acetonitrile derivatives (6 or 7). It is important to mention that there are several methods for displacement of nitro groups which use dipolar aprotic solvents to attain a high yield of ether groups15, 16. In this study the estrone and pregnenolone were reacted with 3 in medium basic to form 6 or 7. The 1H NMR spectrum of 6showed several signals at 1.00 ppm for methyl group; at 1.22-3.00, 6.78-6.78 and 7.14 ppm for steroid moiety; at 3.66 ppm for methylene group bound to both phenyl and nitrile groups; at 6.98 and 7.36 ppm for phenyl group. The 13C NMR spectrum exhibits the following signals at 13.60 ppm for methyl group; at 21.78, 25.72-114.51, 124.60, 132.61-139.84 and 155.94 ppm for steroid moiety; at 23.50 ppm for methylene group bound to both phenyl and nitrile groups; at 117.10, 122.50, 128.30 and 155.00 ppm for phenyl group; at 117.40 ppm for carbon of nitrilomethane; at 219.80 for ketone group. In addition, the mass spectrum of 6 exhibited molecular ion peak atm/z 385.20.
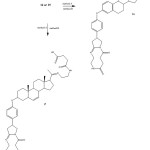 |
Figure 7: Preparation of two pyrrolo-steroid-triazecin-oxobutanoic acids complex (16 or 17) via ciclization of 14 or 15in presence of a carbodiimidederivative (Method A) or boric acid (Method B). |
On the other hand, the 1H NMR spectrum of 7 showed several signals at 0.64 ppm for methyl group bound to steroid nucleus; at 2.10 ppm for methyl bound to ketone; at 1.00-2.04, 4.20-5.40 and 2.20-2.58 ppm for steroid nucleus; at 3.66 ppm for methylene bound to nitrile; at 6.84-7.28 ppm for phenyl group. The 13C NMR spectrum exhibits the following signals at 13.23 ppm for methyl group bound to steroid nucleus; 30.22 ppm for methyl group bound to ketone; 23.42 ppm for methylene bound to nitrile; at 24.00-26.15, 31.40-80.32, 122.24 and 139.00 ppm for steroid moiety; at 115.20, 122.80-127.10 and 156.32 ppm for steroid moiety; at 117.40 ppm for nitrile group; at 209.23 ppm for ketone group. Finally, the mass spectrum of 7 exhibited molecular ion peak atm/z 417.26.
Preparation of steroid-phenyl-pyrrolone derivatives
The second stage involved the preparation of two steroid-phenyl-pyrrolone derivatives (8 and 9). It is important to mention that these compounds were prepared using two methods the first step (Method A) was achieved by reaction of 6 or 7 with with 6-cloro-1-hexyne to form 8 or 9 in mild conditions. The second step (method B) was achieved by the reaction of estrone or pregnenolone with 5 to form 8 or 9. It is important to mention that with the method B the production was higher in comparison with method A. This phenomenon possibly is due to conditions of reaction involved in each method. The 1H NMR spectrum of 8showed several signals at 1.00 ppm for methyl group; at 1.22-3.00, 6.70 and 6.78-7.14 ppm for steroid moiety; at 4.50-5.50 ppm for pyrrole ring; at 6.74 and 7.28 ppm for phenyl group; at 8.01 ppm for amino group. The 13C NMR spectrum exhibits the following signals at 13.60 ppm for methyl group; at 21.78-48.62, 114.26-114.51, 124.60, 130.00 and 132.61-154.82 ppm for steroid moiety; at 50.70, 118.50, 168.60 and 174.38 ppm for pyrrole ring; at 117.58, 128.60, 156.91 ppm for phenyl group; at 219.80 for ketone group. Finally, the mass spectrum of 8 exhibited molecular ion peak atm/z 427.21.
On the other hand, the 1H NMR spectrum of 9 showed several signals at 0.64 and 1.00 ppm for methyl groups bound to steroid nucleus; at 2.10 ppm for methyl group bound to ketone; at 1.04-2.08, 2.20-4.20 and 5.44 ppm for steroid moiety; at 4.56 and 5.50 ppm for pyrrole ring; at 6.84-7.14 ppm for phenyl group; at 8.02 ppm for amino group. The 13C NMR spectrum exhibits the following signals at 13.23 and 19.40 ppm for methyl groups bound to steroid nucleus; at 23.10 ppm for methyl group bound to ketone; at 21.40, 23.98-50.00, 56.84-81.42, 122.04 and 140.00 ppm for steroid moiety; at 50.66, 118.50 and 168.62-174.44 ppm for pyrrole ring; at 114.94, 127.10-128.10 and 156.44 ppm for phenyl group; at 174.44 and 209.23 ppm for ketone groups. In addition, the mass spectrum of 9 exhibited molecular ion peak atm/z 473.29.
Preparation of pyrrol-ethanediamine derivatives
The following stage was achieved by preparation of imino groups involved in the compounds 11 or 12. It is important to mention, that there are several procedures for the synthesis of imines which are described in the literature [15, 16]. For example, the synthesis of imine derivatives by the reaction of the compound 1-[(2-amino-ethylamino)phenyl-methyl]-naphta-len-2-ol with androsterone using as catalyst boric acid [17]. In this study the compounds 11 or 12 were synthesized by the reaction of 8 or 9 using boric acid as catalyst, because it is not an expensive reagent and no special conditions are required for use21. Reaction mechanism involves nucleophilic addition of amino groups to the electron-deficient ketone central carbons resulting imino group. The 1H NMR spectrum of 11showed several signals at 1.22 ppm for methyl bound to steroid nucleus; at 1.22-1.90, 2.08-2.80, 6.68, 6.78 and 7.30 ppm for steroid nucleus; at 3.10, 3.16, 3.50 and 3.66 for methylene groups bound to both imino and amino groups; at 4.90 and 6.70 ppm for pyrrole ring; at 5.30 ppm for amino groups; at 6.74 and 7.24 ppm for phenyl group. The 13C NMR spectrum exhibits the following signals at 15.80 ppm for methyl group; at 22.00-37.56, 41.44-43.32, 54.30, 114.26-114.51, 125.88, 134.14-140.80 and 154.80 ppm for steroid moiety; at 40.94, 41.00, 54.00 and 54.14 ppm for methylene groups bound to both imino and amino groups; at 59.52, 124.20 and 153.32-154.78 ppm for pyrrole ring; at 118.98, 129.10, 130.61 and 158.14 ppm for phenyl group; at 176.80 ppm for imino group. In addition, the mass spectrum of 11 exhibited molecular ion peak atm/z 511.33.
On the other hand, other results showed several signals of 1H NMR spectrum for the compound 12 at 0.90 ppm for methyl group bound to steroid nucleus; at 0.90 and 1.00 ppm for methyl groups bound to steroid nucleus; at 1.80 ppm for methyl group bound to imino; 0.98, 1.22, 1.82-2.50, 4.18 and 5.44 ppm for steroid moiety; at 3.10, 3.16, 3.50 and 3.66 ppm for methylene groups bound to both imino and amino groups; 4.90 and 6.70 ppm for pyrrole ring; at 5.28 ppm for amino groups; at 6.78-7.14 ppm for phenyl group. The 13C NMR spectrum exhibits the following signals at 13.30 and 19.40 ppm for methyl groups bound to steroid nucleus; at 16.70 ppm for methyl group bound to imino; at 21.22-40.00, 42.80, 42.82-50.40, 55.30, 63.10-81.40, 122.11 and 140.02; at 40.94-41.00 and 53.10-54.02 ppm for methylene groups bound to both imino and amin o groups; at 59.52, 124.20, 153.32-154.78 ppm for pyrrole ring; at 116.26, 127.58-128.80 and 157.74 ppm for phenyl group; at 156.70 for imino group. Finally, the mass spectrum of 12 exhibited molecular ion peak atm/z 557.40.
Preparation of carboxy-propionylamino-steroid- succinamic acid
The following stage was achieved by formation of an amide involved in the chemical structure for compounds 14 or 15. It is important to mention that there are many procedures for the formation of amides which are known in the literature, for example the reaction of amine with trichloroisocyanuric acid18, NaH/DMF19, ZrOCl220, tetrabutylammonium bromide21. Analyzing these data and other report which indicate the preparation of amide groups with some carbodiimides22; therefore, in this study 11 or 12 were reacted with succinic acid in presence of a carbodiimide derivative to form the compounds 13 or 14.The 1H NMR spectrum of 14showed several signals at 1.00 ppm for methyl group; at 1.22-2.28, 2.60-2.80 and 6.68-6.78 ppm for steroid moiety; at 7.30 ppm for both amide and carboxyl groups; at 2.50-2.55 for methylene bound to both amide and carboxyl groups; at 3.32-3.70 ppm 55 for methylene bound to both to imino and amide groups; at 4.90-6.70 ppm for pyrrole ring; at 6.72-7.24 ppm for phenyl group. The 13C NMR spectrum exhibits the following signals at 15.80 ppm for methyl group; at 22.00-27.50, 29.76, 32.42, 31.56-43.32, 54.30, 114.26-114.51, 125.88, 134.14-140.80 and 154.78 ppm for steroid moiety; at 28.84-30.00 ppm for methylene groups bound to both amide and carboxyl groups; at 36.00-36.08 and 54.10-54.24 ppm for methylene groups bound to both imino and amide groups; at 59.52, 124.20 and 153.32-154.78 ppm for pyrrole ring; at 118.98, 129.10-130.61 and 158.14 ppm for phenyl group; at 166.80 ppm for amide groups; at 173.56 ppm for imino groups; at 176.88 ppm for carboxyl groups. In addition, the mass spectrum of 14 exhibited molecular ion peak atm/z 711.36.
The 1H NMR spectrum of 15showed several signals at 0.78 and 0.90 ppm for methyl group bound to steroid nucleus; at 1.68 ppm for methyl group bound to imino group; at 1.30-1.56, 1.70-2.42, 4.00, 5.40-5.60 ppm for steroid moiety; at 2.50-2.56 ppm for methylene group bound to both amide and carboxyl groups; at 3.46- 3.74 ppm for methylene groups bound to both imino and amide groups; at 4.88 and 6.70 ppm for pyrrole ring; at 6.80-7.12 ppm for phenyl groups; at 8.14 ppm for both amide and carboxyl groups. The 13C NMR spectrum exhibits the following signals at 14.64 and 16.36 ppm for methyl groups bound to steroid nucleus; at 22.58 ppm for methyl group bound to imino group; at 21.22, 26.20-28.90, 34.08-35.00, 37.46-53.00, 57.00, 82.66 and 129.48-130.80 ppm for steroid moiety; at 28.90-30.00 ppm for methylene groups bound to both amide and carboxyl groups; at 35.70-36.00 and 54.10 ppm for methylene groups bound to both imino and amide groups; at 59.32, 124.21 and 153.32-154.78 ppm for pyrrole ring; at 116.00, 127.58-128.80 and 157.40 ppm for phenyl groups; at 162.70 ppm for nitrile group; at 166.80 ppm for amide group; at 173.64 ppm for carboxyl group. In addition, the mass spectrum of 15 exhibited molecular ion peak atm/z 743.42.
Preparation of pyrrolo-steroid-triazecin-oxobutanoic acids complex
Several triazecinone derivatives have been prepared using different reagents such as 1,2diaminoethane, DMAP, tetrachloro-2-azabuta-1,3-dienes23-25 and others. In this study, the synthesis of steroid-pyrrolo-triazecin derivatives (16 or 17) was via cyclization of 14 or 15 using a carbodiimide derivative (method B) or boric acid (method A). It is important to mention with method A the yielding was higher in comparison with Method B, this phenomenon possibly is result of the reaction conditions that are involved in this study or to different structure chemical of catalyzers.
The 1H NMR spectrum of 16 showed several signals at 1.02 ppm for methyl group; at 1.22-2.18, 2.25-2.28, 2.60-2.80, 6.68,6.76 and 7.28 ppm for steroid moiety; at 2.22, 2.40, 3.52, 3.88 ppm for methylene groups involved in the triazecine-dione ring; at 2.52-2.56 ppm for methylene groups bound to both amide and carboxyl groups; at 3.32 and 3.55 ppm for methylene groups bound to both imino and amide groups; at 4.88-5.16 and 7.24 ppm for pyrrole ring; at 6.75 and 7.29 ppm for phenyl group; at 7.76 ppm for amide involved in the triazecine-dione ring; at 8.58 ppm for both amide and carboxyl groups involved in the arm bound to D-ring of steroid nucleus. The 13C NMR spectrum exhibits the following signals at 15.80 ppm for methyl group; at 22.00-27.50, 29.76, 32.42, 37.60, 41.44-43.32, 54.30, 114.26, 114.51, 125.88, 134.14-140.80 and 154.78 ppm for steroid moiety; at 28.84 and 30.00 for methylene groups bound to both amide and carboxyl group; at 31.60, 33.50, 37.79 and, 62.12 ppm for methylene groups involved in the triazecine-dione ring; 36.08 and 54.24 ppm for methylene groups bound to both amide and imino groups; at 58.92, 124.90 and 147.50-150.66 ppm for pyrrole ring; at 119.20, 129.10, 130.50 and 158.14 ppm for phenyl group; at 166.80-170.60 for amide groups; at 173.56 ppm for carboxyl group; at 176.88 ppm for imino group; at 180.94 ppm for ketone group. In addition, the mass spectrum of 16 exhibited molecular ion peak atm/z 693.35.
The 1H NMR spectrum of 17 showed several signals at 0.98 and 1.00 ppm for methyl groups bound to steroid nucleus; at 1.28-1.56, 1.70-2.14, 2.30-2.40, 4.00 and 5.36-5.58 ppm for steroid moiety; at 2.20, 2.42, 3.58 and 3.96 ppm for methylene groups involved in the triazecine-dione ring; at 2.50-2.56 ppm for methylene groups bound do both amide and carboxyl groups; at 3.48 and 3.62 ppm for methylene groups bound to both imino and amide groups; at 3.80, 3.98 and 4.22 ppm for pyrrole ring; at 6.80-7.00 ppm for phenyl group; at 7.74 ppm for amide involved in the triazecine-dione ring; at 7.90 ppm for both amide and carboxyl groups involved in the arm bound to D-ring of steroid nucleus. The 13C NMR spectrum exhibits the following signals at 14.60 and 16.30 ppm for methyl groups bound to steroid nucleus; at 22.60 ppm for methyl group bound to imo group; at 21.22, 26.20-26.25, 28.90, 34.08-35.00, 37.40-37.94, 43.60-51.00, 53.00, 57.00, 82.70 and 129.44-130.80 ppm for steroid moiety; at 28.98 and 30.00 ppm for methylene groups bound to both amide and carboxyl groups; at 35.70 and 53.32 ppm for methylene groups bound to both imino and amide groups; at 31.60-33.50, 37.98 and 62.16 ppm for methylene groups involved in the triazecine-dione ring; at 43.36-43.40, 51.70 and 156.30 ppm for pyrrole ring; at 114.54-128.74, and 156.08 ppm for phenyl group; at 162.66 ppm for imino group; at 166.80-170.60 ppm for amide groups; at 173.64 for carboxyl group; at 180.98 for ketone group. In addition, the mass spectrum of 17 exhibited molecular ion peak atm/z 693.35.
Conclusions
In this study is reported a straightforward route for synthesis of two steroid-pyrrolo-triazecin derivatives. The proposed methods offer some advantages such as simple procedure, low cost, and ease of workup.In addition, it is also important to mention that these compounds could be used as pharmacological tools to evaluate the biological activity from other type of steroid derivatives.
References
- Schley, N.; Dobereiner, E; CrabtreeH. Organometallics. 2011, 30, 4174-4179.
CrossRef - Zhang, M; Neumann, H; Beller, M. Angew. Chem. 2013, 125, 625-6.29.
- Kanchithalaivan, S; Kumar, R; Perumal, S;Steroids. 2013, 78, 409-417.
CrossRef - Crawley, M;Goljer, I;Jenkins, D;Mehlmann, J;Nogle, L; Dooley, R;Mahaney, P.Org. Lett.2006, 8, 5837-5840.
CrossRef - Saito, A;Konishi, T;Hanzawa, Y.Org. Lett.2010, 12, 372-374.
CrossRef - Yu, M;Pagenkopf, B.Org. Lett.2003, 5, 5099-5101.
CrossRef - Galliford, C;Scheidt, K.J. Org. Chem.2007, 72, 1811-1813.
CrossRef - Davies, P; Martin, N.Org. Lett.2009, 11, 2293-2296.
CrossRef - Zhao, X;Zhang, E;Tu, Y;Zhang, Y;Yuan, D;Cao, K.Org. Lett.2009, 11,4002-4004.
CrossRef - Cyr, D;Martin, N; Arndtsen,B.Org. Lett. 2007, 9, 449-452.
CrossRef - Minetto, G;Raveglia, L;Taddei, M.Org. Lett.2004, 6,389-392.
CrossRef - Murugesan, D;Mital, A; Kaiser, M;Shackleford, D;Morizzi, J;Katneni, K; Campbell, M.J. Med. Chem.2013, 56, 2975-2990.
CrossRef - Imhof, W; Berger, D;Kötteritzsch, M;Rost, M;Schönecker, B. Adv. Synth. Cat.2001, 343,795-801.
CrossRef - Ahmad,A; Husain, A; Khan, S;Mujeeb,A. J. Saudi Chem. Soc.2015, 19,340-346.
CrossRef - Shirayev, A;Moiseev, I;Karpeev, S;Arkivok. 2005, iv, 199-207.
- Uppiah, D; Bhowon, M; Jhaumeer, S. E-J. Chem. 2009, 6, S195-200.
- Figueroa-Valverde, L; Díaz-Cedillo, F; García-Cervera, E; Rosas-Nexticapan, M; Ramos-López, M.Asian J. Chem. 2013, 25, 6724-6726.
- Hiegel,G;Hogenauer,T; Lewis, J.Syn. Comm. 2005, 35,2099-2105.
CrossRef - Song, E; Kaur, N; Park, M; Jin, Y; Lee, K.Eur. J. Med. Chem.2008, 43,1519-1524.
CrossRef - Reddy, C; Nagaraj, A; Jalapathi, P.Chinese Chem. Lett.2007, 18,1213-1217.
CrossRef - Meghdadi, S; Mirkhani, V; Ford, P.Synt. Org. Chem. 2012, 42, 246-250.
CrossRef - Figueroa-Valverde, L; Díaz-Cedillo, F; García-Cervera, E; Pool-Gómez, E; López-Ramos, M. BulgarianChem. Comm. 2013, 45, 71-76.
- Mokrov, G;Likhosherstov, A;Lezina, V;Gudasheva, T; Bushmarin, I; Yu, M.Russian Chem. Bull.2011, 60,1694-1702.
CrossRef - Burnett, N;Hosmane, R. Tetrahedron. 2002, 58, 9567-9578.
CrossRef - Snieckus, V; Macklin, T.Synfacts.2006, 10, 998-998.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


