Reverse docking study unravels the potential Mycobacterium tuberculosis enzyme targets of Agelasine F
Junie B. Billones
Department of Physical Sciences and Mathematics, College of Arts and Sciences, and Institute of Pharmaceutical Sciences, National Institutes of Health
University of the Philippines Manila Taft Avenue, Ermita, Manila, Philippines 1000.
Corresponding Author Email: jbbillones@up.edu.ph
DOI : http://dx.doi.org/10.13005/ojc/320210
Article Received on :
Article Accepted on :
Article Published : 09 May 2016
The natural product Agelasine F found in marine sponge Agelas sp. along with the known inhibitors of the target enzymes in Mycobacterium tuberculosis (Mtb), and the first-line tuberculosis (TB) drugs have been docked to enoyl reductase (InhA), Isoniazid-resistant I21V and S94A enoyl-ACP (COA) reductase mutant enzymes, 7,8-diaminopelargonic acid synthase (DAPAS), pantothenate synthetase (PS), and lipoate-protein ligase B (LipB) enzymes. Among the compounds studied, Agelasine F came out as the best inhibitor for InhA, S94A enoyl-ACP, and DAPAS. Except for LipB, Agelasine F exhibited superior binding affinity compared to the known inhibitors of the studied Mtb enzyme targets. Moreover, Agelasine F possesses remarkable enzyme inhibitory potential and drug-like properties.
KEYWORDS:Agelasine F; molecular docking; Mycobacterium tuberculosis; enoyl reductase (InhA); 7;8-diaminopelargonic acid synthase (DAPAS); pantothenate synthetase (PS); lipoate-protein ligase B (LipB)
Download this article as:| Copy the following to cite this article: Billones J. B. Reverse docking study unravels the potential Mycobacterium tuberculosis enzyme targets of Agelasine F. Orient J Chem 2016;32(2) |
| Copy the following to cite this URL: Billones J. B. Reverse docking study unravels the potential Mycobacterium tuberculosis enzyme targets of Agelasine F. Orient J Chem 2016;32(2). Available from: http://www.orientjchem.org/?p=15925 |
Introduction
Tuberculosis (TB) is an airborne infectious disease caused by Mycobacterium tuberculosis. According to World Health Organization (WHO), the estimated number of new TB cases in 2014 was almost 9.6 million with 1.5 million TB deaths worldwide, including 400,000 deaths among HIV-positive people.1 In 2014, there were approximately 480,000 new cases of multidrug resistant TB (MDR-TB) and an estimated 190,000 deaths from MDR-TB. By 2015, 105 countries had reported at least one extensively drug resistant XDR-TB case. Among the six WHO regions, South-East Asia had the highest TB incidence in 2014. In the same year, the Philippines was ranked 8th among countries most afflicted with TB.1 TB is the 6th leading cause of mortality in the Philippines.2
Although drugs have been developed to combat the disease, the emergence of drug resistant strains of M. tuberculosis make the campaign for successful treatment of tuberculosis more difficult.3 The rapid emergence of MDR-TB and XDR-TB has been considered a public health emergency and undermines the Millennium Development Goal of reducing TB incidence to 50% by 2015.4 Thus, there is an urgent need to develop new therapeutics against TB.
Drug discovery efforts today have been taking advantage of computer-based technologies in order to fast track the intricate stages in drug discovery pipeline. In particular, molecular docking has been commonly applied in screening molecular databases against a putative druggable macromolecular target.5 Interestingly, it has also been employed recently in screening a database of druggable targets against certain drug-like molecules.6 The latter approach is called reverse or inverse docking.7,8
Reverse docking has been employed in this study to furnish the first principles of bioactivity and substantiate the earlier accounts, which demonstrate the antimicrobial9 and particularly antimycobacterial10 activity of Agelasine F, a secondary metabolite isolated from a marine sponge (Agelas sp).9,11,12 The six enzymes that were screened against Agelasine F included the isoniazid (INH) targets: enoyl reductase (InhA), and I21V and S94A enoyl-ACP (COA) reductase mutants; and other druggable targets such as pantothenate synthetase (PS), lipoate-protein ligase B (LipB), and 7,8-diaminopelargonic acid synthase (DAPAS).
Experimental
The three-dimensional (3D) structural data of the target enzyme were retrieved as pdb file from the Protein Data Bank (PDB) (www.rcsb.org) while the structure of ligand molecules were obtained from PubChem (pubchem.ncbi.nlm.nih.gov) or inputted using the drawing tool of DockingServer and stored as sdf file. All computational works were performed using MacBook Air running on Mac OSX 10.8 platform.
Docking calculations were carried out using DockingServer.13 The MMFF94 force field14 was used for energy minimization of ligand molecule using DockingServer. Gasteiger partial charges were added to the ligand atoms. Non-polar hydrogen atoms were merged, and rotatable bonds were defined. Essential hydrogen atoms, Kollman united atom type charges, and solvation parameters were added with the aid of AutoDock tools.15 Affinity (grid) maps of 20×20×20 Å grid points and 0.375 Å spacing were generated using the Autogrid program. AutoDock parameter set- and distance-dependent dielectric functions were used in the calculation of the van der Waals and the electrostatic terms, respectively. Docking simulations were performed using the Lamarckian genetic algorithm (LGA) and the Solis & Wets local search method.16 Initial position, orientation, and torsions of the ligand molecules were set randomly. Typically, each docking experiment was derived from 10 different runs that were set to terminate after a maximum of 250000 energy evaluations. The population size was set to 150. During the search, a translational step of 0.2 Å, and quaternion and torsion steps of 5 were applied.
Results and Discussion
The three-dimensional structures of some Mtb drug targets are shown in Fig. 1. The druggability of the enzyme targets studied here has been well established and demonstrated in numerous studies. Specifically, the enzyme enoyl-acyl carrier protein (ACP) reductase (PDB ID: 2B35) also called InhA17, with mutants (i.e. I21V, PDB ID: 2IE0 and S94A, PDB ID: 2IEB), is the target of isoniazid (INH)18, a first-line anti-TB drug. InhA inhibitors like INH prevent the biosynthesis of mycolic acid,
 |
Figure 1: Ribbon representation of the structure of Mycobacterium tuberculosis enzyme targets. 2B35 – enoyl (InhA) reductase17; 2IE0 – I21V enoyl-ACP (COA) reductase mutant; 2IEB – S94A enoyl-ACP (COA) reductase mutant, 1N2H – pantothenate synthetase (PS); 1W66 – lipoate-protein ligase B (LipB); 3LV2 – 7,8-diaminopelargonic acid synthase (DAPAS) obtained from PDB (www.rcsb.org). Click here to View figure |
which largely compose the mycobacterial cell wall. Pantothenate synthetase (PS) is another attractive Mtb target. It serves to catalyze the production of pantothenate (Vitamin B5), a precursor for the biosynthesis of coenzyme A (CoA) and acyl carrier protein (ACP)19, which are essential for the cell’s metabolism. Since humans lack PS, this enzyme is an attractive target for antitubercular drugs.20 Lipoate-protein ligase B (LipB), also known as octanoyltransferase, catalyzes the first committed step in the biosynthesis of lipoyl cofactor, which is essential for the function of several key enzymes involved in the oxidative metabolism of M. tuberculosis.21 Moreover, a relatively new putative Mtb target, 7,8-diaminopelargonic acid synthase (DAPAS), the second enzyme in the biotin biosynthetic pathway, is necessary for de novo biotin
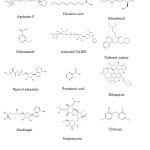 |
Figure 2: Chemical structures of inhibitors of druggable Mycobacterium tuberculosis enzyme targets. Click here to View figure |
biosynthesis.22 It has been shown that the disruption of the Mtb biotin biosynthesis machinery results in rapid bacterial clearance.23 Incidentally, we have performed virtual screening and molecular docking studies on some of these druggable targets in Mtb such PS24,25, LipB26 and DAPAS27.
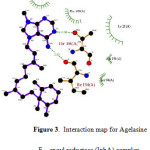 |
Figure 3: Interaction map for Agelasine F – enoyl reductase (InhA) complex. Click here to View figure |
The results of molecular docking studies are presented in Table 1. Although the main focus of the study was Agelasine F, the other known inhibitors including some TB drugs (Fig. 2) were also docked to the selected Mtb enzyme targets for comparison. It is very interesting to note that Agelasine F showed better binding affinity with InhA compared with the active form of isoniazid, a first-line TB drug. Agelasine was observed to form H-bonding with ILE194 and THR196, and hydrophobic interaction with TYR158, PHE149, ILE21, ALA191 and SER94 (Fig. 3). Nonetheless, Rifampicin turned out to be slightly better than Agelasine F in inhibiting the native InhA enzyme. On the other hand, Agelasine F outperformed all the other compounds in inhibiting S94A enoyl-ACP (COA) reductase mutant, albeit it only ranks 3rd against the I21V mutant, behind Ciprofloxacin and Triclosan. However, in a 100-run calculation, one binding conformation of Agelasine F showed better binding energy (-9.57 kcl/mol) than Ciprofloxacin (-7.74 kcal/mol) and Triclosan (-7.55 kcal/mol).
Table 1: Estimated Binding Energies (kcal/mol) Using DockingServer for Inhibitors of Druggable Mycobacterium tuberculosis Enzyme Targets.
| Compound |
Estimated Binding Energy (kcal/mol) |
|||||
|
2B35 |
2IE0 |
2IEB |
1N2H |
1W66 |
3LV2 |
|
| Agelasine F |
-6.89 |
-7.55 |
-8.16 |
-7.05 |
-3.76 |
-7.21 |
| Ciprofloxacin |
-6.82 |
-7.58 |
-7.51 |
-7.44 |
-5.59 |
-6.54 |
| Decanoic acidc | -3.27 |
-4.12 |
-4.55 |
-3.94 |
-4.09 |
-3.47 |
| Ethambutol |
-2.47 |
-2.99 |
-3.19 |
-3.71 |
-1.15 |
-5.69 |
| Ethionamide |
-5.04 |
-5.83 |
-5.34 |
-5.92 |
-5.53 |
-5.17 |
| Isoniazid-NADa |
-1.85 |
0.62 |
2.80 |
30.08 |
15.53 |
3.32 |
| Nafronyl oxalateb |
-6.10 |
-6.46 |
-6.80 |
-7.00 |
-7.16 |
-5.61 |
| Pantoyl adenylateb |
-2.61 |
-3.36 |
-0.94 |
-2.93 |
-2.36 |
-0.74 |
| Pyrazinoic acid |
-4.15 |
-4.56 |
-4.53 |
-4.47 |
-4.69 |
-4.57 |
| Rifampicin |
-8.36 |
-6.80 |
-7.02 |
-7.54 |
-3.3 |
-6.48 |
| Sinefungind |
-5.64 |
-5.72 |
-6.18 | -6.26 |
-4.27 |
-6.30 |
| Streptomycin |
-5.65 |
-5.67 | -3.71 |
-1.23 |
9.10 |
-4.92 |
| Triclosan |
-6.87 |
-7.76 |
-7.53 |
-7.34 |
-6.10 |
-6.08 |
a known inhibitor of InhA (2B35, 2IE0, 2IEB), b known inhibitor of PS (1N2H), c known inhibitor of LipB (1W66), d known inhibitor of DAPAS (3LV2)
Against pantothenate synthetase (PS), the binding energy of Agelasine F is comparable to that of Ciprofloxacin, Rifampicin, and Triclosan, and better (more negative) than the known inhibitors, nafronyl oxalate and pantoyl adenylate. In fact, the binding energy of Agelasine F (-8.83 kcal/mol) was even better than Ciprofloxacin (-7.63 kcal/mol) and Triclosan (-7.97 kcal/mol) in a more elaborate 100-run experiment. On the other hand, Agelasine F is not a good inhibitor of LipB as its binding energy is slightly lower than that of the substrate decanoic acid and at least 4 kcal/mol smaller than nafronyl oxalate, the best ligand in the group.
Agelasine F demonstrated excellent binding affinity against DAPAS, its binding energy being the largest among the compounds studied. Fig. 4 provides some insight on the binding interaction of Agelasine F with DAPAS. Evidently, Agelasine F forms hydrophobic interaction with TRP64, PHE402, TYR157, TRP65, ALA226, TYR407, and in various ways with LYS283, THR66, and ARG400 of DAPAS.
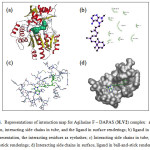 |
Figure 4: Representations of interaction map for Agilasine F – DAPAS (3LV2) complex: a) protein in cartoon, interacting side chains in tube, and the ligand in surface renderings; b) ligand in ball-and-stick representation, the interacting residues as eyelashes; c) Interacting side chains in tube, ligand in ball-and-stick renderings; d) Interacting side chains in surface, ligand in ball-and-stick renderings. Click here to View figure |
Given the favorable in silico binding characteristics of Agelasine F with several MTB drug targets, it is instructive to calculate the relevant drug properties of this natural compound as well. Accordingly, the following druglikeness indicators for Agelasine F were obtained by the use of online program Molinspiration Property Engine v2011.04 (http://www.molinspiration.com/cgi-bin/properties): milogP = 2.544, tPSA = 60.626 Å2, MW = 422.641, NON = 5, NOHNH = 2, Nviol = 0, Nrot= 8, Vol = 437.172 Å3 (Table 2).
Table 2: Calculated Propertiesa Related to Druglikeness for the Inhibitors of Mycobacterium tuberculosis Enzyme Targets.
|
Compound |
logP |
tPSA |
MW |
No. of H-Bond donors |
No. of H-Bond acceptors |
Enzyme Inhibitor Score |
| Agelasine F |
2.54 |
60.63 |
422.6 |
2 |
5 |
+0.47 |
| Decanoic acid |
4.03 |
37.3 |
172.3 |
1 |
2 |
-0.07 |
| Ethambutol |
0.35 |
64.5 |
204.3 |
4 |
4 |
-0.08 |
| Ethionamide |
1.46 |
38.9 |
166.2 |
2 |
2 |
-0.53 |
| Isoniazid |
-0.97 |
68.0 |
137.1 |
3 |
4 |
-0.66 |
| Nafronyl oxalate |
4.94 |
38.8 |
383.5 |
0 |
4 |
+0.03 |
| Pantoyl adenylate |
-1.49 |
232.6 |
477.4 |
7 |
15 |
+1.32 |
| Pyrazinamide |
-0.71 |
68.9 |
123.1 |
2 |
4 |
-1.43 |
| Rifampicin |
2.62 |
220.2 |
823.0 |
6 |
16 |
-2.42 |
| Sinefungin |
-3.89 |
208.7 |
381.4 |
9 |
12 |
+1.14 |
| Streptomycin |
-4.32 |
316.2 |
579.6 |
15 |
18 |
+0.37 |
| Triclosan |
5.13 |
29.5 |
289.5 |
1 |
2 |
-0.01 |
aCalculated using Molinspiration v2011.06 (http://www.molinspiration.com/cgi-bin/properties)
The property milogP is the Molinspiration calculated logarithm of the partition coefficient in biphasic octanol-water mixture. The logP has been commonly used as estimate of the hydrophobicity of a molecule,28 a key factor in drug transport and permeability, hydrophobic interactions, metabolism and toxicity.29-31 The topologic polar surface area (tPSA), defined as the sum of surfaces of polar atoms, has also been shown to influence drug transport,32 absorption, bioavailability, and blood-brain barrier penetration.33 The MW and Vol are simply the weight and volume of the molecule while NON, NOHNH, Nviol, Nrot, and Natom are the number of H-bond acceptors, H-bond donors, rotatable bonds, and atoms in the molecule, respectively.
It is very encouraging to find that the critical numbers for druglikeness like the Lipinski’s “Rule of Five” (i.e. logP < 5, MW < 500, NON < 10, NOHNH < 5),34 were satisfactorily met by Agelasine F. Moreover, the polar surface area of Agelasine F indicates good cell permeability, the critical value being 140 Å2, beyond which the cell membrane permeation is rather poor.33 The number of rotatable bonds (Nrot) is a measure of molecular flexibility, which partly affects cell membrane permeability.35-36 The optimal value for Nrot is ≥7, albeit Nrot ≥ 11 is the critical limit and coded red or Traffic Light (TL) 3 in Lobell scoring system for oral bioavailability.37 Nevertheless, although coded yellow or TL2 in molecular flexibility scoring system, the eight rotatable bonds in Agelasine F are well within the threshold level.
 |
Figure 5: Calculated distribution of activity score for enzyme inhibitors compared with scores for about 100,000 average drug-like molecules; reconstructed from Molinspiration (www.molinspiration.com/docu/miscreen/druglikeness.html). The enzyme inhibitor score for inhibitors of druggable Mtb targets are estimated by their locations in the plot. Click here to View figure |
Finally, it is also instructive to predict the overall enzyme inhibitory potential of Agelasine F. Thus, the enzyme inhibitor score for Agelasine F was calculated using Molinspiration Bioactivity Score v2011.06. Fig. 5 shows the distribution of activity scores for enzyme inhibitors compared with those for “average drug-like molecules”. The score allows straightforward separation of active and inactive molecules. The larger the score value is, the higher is the chance that the particular molecule will be active. The enzyme inhibitor score of 0.47 for Agelasine F places it above average among active enzyme inhibitors. Moreover, it outscored the compounds studied here, although it is overtaken only by pantoyl adenylate, the natural ligand for the enzyme pantothenate
Conclusion
Agelasine F, a known bioactive compound against Mycobacterium tuberculosis has been successfully docked to several druggable Mtb enzyme targets, namely, enoyl reductase (InhA), Isoniazid-resistant I21V and S94A enoyl-ACP (COA) reductase mutant enzymes, 7,8-diaminopelargonic acid synthase (DAPAS), pantothenate synthetase (PS), and lipoate-protein ligase B (lipB) enzymes. The computed binding affinities of Agelasine F vis-à-vis the known inhibitors of putative drug targets and selected TB drugs were highly promising. In particular, among the compounds studied, Agelasine F turned out to be the best inhibitor for InhA, S94A enoyl-ACP, and DAPAS. Moreover, Agelasine F satisfies the criteria of druglikeness such as Lipinski’s rule, polar surface area, number of rotatable bonds, and enzyme inhibitory potential. These results should encourage the development of a new class of antitubercular agents based on Agelasine F architecture.
Acknowledgement
This project is supported by PHAREX research grant through the National Institutes of Health-Philippines (NIH 13-0004), University of the Philippines Manila. JBB also appreciates the Gawad Sentenaryo 2015 awarded to him by UP Manila.
References
- WHO. World Health Organization Global Tuberculosis Report 2015. Available from: http://apps.who.int/iris/bitstream/10665/191102/1/9789241565059_eng.pdf.
- Department of Health (DOH), Republic of the Philippines, Leading Causes of Mortality (n.d.). (Retrieved from http://www.doh.gov.ph/node/198.html).
- Corbett. L. and Raviglione. M., Global Burden of Tuberculosis: Past, Present and Future, in: Tuberculosis and the Tubercle Bacillus, S. T. Cole, (eds.), ASM Press, Washington D.C. 2005,. 3-12.
CrossRef - US Agency for International Development, in: Accelerating Impact: Expanding Access to Care, Washington, DC. Available from: https://www.usaid.gov/sites/default/files/documents/1864/tb_report 2013.pdf.
- Morris. G. M. and Wilbey. M.L., Molecular Docking, in: Molecular Modeling of Proteins, Methods in Molecular Biology, A. Kukol (ed.), Humana Press, New Jersey 2008,. 443,. 365 – 382.
- Zheng. R., , Chen. T, Lu. T. , Int. J. Mol. Sci., 2011.,12, 5200- 5212.
- Chen. Y. Z. and. Ung C. Y., J. Mol. Graph. Model., 2001. 20(3), 199-218
- Li. H., Gao. Z., Kang. L., Nucleic Acids Res., 2006. 34, 219- 224
- Capon. R. J., Faulkner. D. J., J. Am. Chem. Soc., 1984,106, 1819-1922.
- Mangalindan. G. C., Talaue. M. T., Cruz. L.J., Planta Med., 2000.66(4), 364- 365
- Wu. H., Nakamura. H., Kobayashi. J., Bull. Chem. Soc. Jpn., 1986 59, 2495- 2504.
- Fu. X.,. Schmitz. F. J, Tanner. R. S.,, J. Nat. Prod., 1998.61, 548-550
- Bikadi. Z. and Hazai. E., J. Cheminf., 2009.1, 15
- Halgren. T. A., J. Comp. Chem., 1998.,17(5-6), 490 – 519
- Morris. G. M., Goodsell. D.S., Halliday. R.S, J. Comp. Chem., 1998.19(14), 1639- 1662
- Solis. F. J. and Wets. R. J., Math. Oper. Res., 1981., 6(1), 19- 30.
- Sullivan. T. J., Truglio. J. J., Boyne. M. E., ACS Chem. Biol., 2006.1(1), 43- 53
- Banerjee. A., Dubnau. E., Quémard. A., Science, 1994.,263, 227- 230 .
- Zheng. R. and Blanchard. J. S., Biochemistry, 2001.,40(43), 12904- 12912 .
- White. E. L., Southworth. K., Ross. L.,, J. Biomol. Screen., 2007.12(1), 100- 105
- Ma. Q., Zhao. X., Eddine. A., Proc. Natl. Acad. Sci. U S A, 2006.,103(23), 8662- 8667 .
- Keer. J., Smeulders. M. J.,. Gray. K. M, Microbiology, 2000.,146(9), 2209- 2217.
- Sassetti. C. M. and Rubin. E. J., Proc. Natl. Acad. Sci. U S A, 2003; 100(22):12989–12994
- Uy. V. C. C, and Billones. J. B., Phil. Sci. Lett., 2012.5(2), 122- 130
- Yang. C. T. M. and Billones. J. B., Phil. J. Sci., 2012.141(2), 187 – 196
- Billones, J.B.; Carrillo, M.C.O.; Organo, V.G.; Macalino, S.J.Y.; Emnacen, I.A.; Sy. J.B.A. Orient. J. Chem., 2013, 29(4), 1457 – 1468.
CrossRef - Billones. J. B., Carrillo. M. C. O., Organo. V. G., Macalino. S. J. Y., Emnacen. I. A., Sy. J. B. A. Curr. Enzyme Inhibit., 2014.10, 105 – 112
- Sampaco, A.R.B. III; Billones, J.B. Orient. J. Chem., 2015, 31(4), 1859 – 1865
CrossRef - Valle, A.M.F; Billones, J.B. Orient. J. Chem., 2014, 30(3), 1137 – 1145.
CrossRef - Fujita. T., Iwasa. J., Hansch. C., J. Am. Chem. Soc., 1964.,86(23), 5175- 5180.
- Fujikawa. M., Ano. R., Nakao. K.,., Bioorg. Med. Chem., 2005. 13(15), 4721- 4732
- Yang. S., Bumgarner. J. G., Kruk. L. F.,., J. Chromatogr. A, 1996. 721, 323- 335
- Kubinyi. H., Farmaco Sci., 1979.34(3), 248- 276
- Fernandes. J. and Gattass. C. R., J. Med. Chem., 2009.,52(4), 1214 -1218 .
- Ertl. P., Rohde. B., Selzer. P., J. Med. Chem. 2000. 43, 3714-3717
- Lipinski. C. A., Lombardo. F., Dominy. B. W. ,. Adv. Drug Deliv. Rev., (2001).,46(1-3), 3 – 26
- Veber. D. F., Johnson. S. R., Cheng. H. Y., J. Med. Chem., 2002,.45(12), 2615-2623
- Vistoli. G., Pedretti. A.,. Testa. B, Drug Discov. Today, 2008.13(7-8), 285- 294
- Lobell. M., Hendrix. M,. Hinzen. B,., Chem.Med.Chem., 2006. 1(11), 1229- 1236

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


