New Cerium(III) Complex Of Schiff Base (E)-N-Benzylidene-4-Methoxyaniline: Synthesis And Density Functional Theoretical Study Of Vibrational Spectra
Imanelakehal1∗, Farhihalamia1, Saoussenlakehal2, Abbas Boukhari3 and Abdelha Fiddjerourou1
1Organic Synthesis and Biocatalys is Laboratory,Faculty of Science, Chemistry Department, P.B.:12,23000,Annaba,Algeria.
2Applied Organic Chemistry Laboratory,Faculty of Science, Chemistry Department,P.B.:12,23000,Annaba,Algeria.
3Organic Synthesis Modeling and Optimization of Chemical Processes Laboratory, Faculty of Science,Chemistry Department, P.B.:12,23000,Annaba,Algeria
Corresponding author emai; lakehal.imane.12@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/320216
Article Received on :
Article Accepted on :
Article Published : 12 Apr 2016
Complex of cerium (III) with (E)-N-benzylidene-4-methoxyaniline is synthesized through a one-pot three-componentreaction from aromatical dehyde, aromatic amine and the CeCl3· 7 H2O, as an efficient catalyst. This cerium(III)com- plexis characterized by IR, 1H, and 13 CNMR-spectros copy and mass-spectraldata. Molecular structure, Mullikan charges, thermodynamic parameters; vibrational frequencies and intensities were calculated by Density Functional theory methods (B3LYP,B3PW91,mPW1PW91 and PBEPBE) using the SDD basis set. The comparison between the calculated and experimental data inorder to attain the best quality and to predict the structure, the best perfor-mance in the vibration spectra perfected of the title compound,we have found that the harmonic vibration computed are in a good agreement with the observed in IRspectrum, for closest match we calculated optimal scaling factors can recommend for the IR spectral future predictions for unknown compounds of this class. In order to better comparison, we also root meansqu are values of the experimental and calculated IR bands are 16.64,16.64,17.45,17.66 and the mean absolute percentage error values are1.33,1.39,1.40, and 1.5forB3LYP, B3PW91,mPW1PW91, and PBEPBE methods respectively
KEYWORDS:imine complexed; CeCl3,DFT; vibrational frequencies
Download this article as:| Copy the following to cite this article: Imanelakehal, Farhihalamia, Saoussenlakehal, Boukhari A, Fiddjerourou A. New Cerium(III) Complex of Schiff Base (E)-N-Benzylidene-4-Methoxyaniline: Synthesis And Density Functional Theoretical Study of Vibrational Spectra. Orient J Chem 2016;32(2). |
| Copy the following to cite this URL: Imanelakehal, Farhihalamia, Saoussenlakehal, Boukhari A, Fiddjerourou A. New Cerium(III) Complex of Schiff Base (E)-N-Benzylidene-4-Methoxyaniline: Synthesis And Density Functional Theoretical Study of Vibrational Spectra. Orient J Chem 2016;32(2). Available from: http://www.orientjchem.org/?p=15267 |
Introduction
Schiff bases [1–7] and their metal complexes represent one of the most areas of material science, catalysis and chemical research. [8–21] The efficiency of these com- pounds often enhanced upon coordination to a metal has been proven for the drug action acceleration of therapeutic agents [22–26] and for multi-compounds re- action where these complexes are used as intermediates. Recently, extensive research has surrounded the synthesis of lanthanide complexes and precisely the cerium (III) complexes using cerium (III) heptahydrate
(CeCl3 · 7 H2O)[27].
This compounds are prepared by a reaction of a primary amine with an aldehyde and CeCl3 • 7 H2O as a catalyst (Figure 1). In this present work a cerium (III) complex of new Schiff base (E)-N-benzylidene-4- methoxyaniline noted (P2-CeCl3) has been synthesized and characterized by elemental analysis, IR, NMR and Mass spectral data.[28–32].
To our best knowledge no structural data and detailed interpretation of the vibrational spectra of the title com- pound are mentioned in the literature, this prompted us to look into the vibrational spectroscopy of this molecule more carefully. Therefore, this theoretical study aimed to determine molecular and chemical properties of (P2-CeCl3).
An comprehensive theoretical study carried out for predict the optimized geometric parameters, the Mullikan charges, thermodynamic parameters and vibrational frequencies, involving the cerium (III). The Stuttgrat Dresden (SDD) effective core potential [33,34] basis set was used with four DFT methods including B3LYP, B3PW91, mPW1PW91 and PBEPBE methods. For a comparative performance of these four different DFT methods, commit to the detailed theoretical and experimental investigation of the vibrational spectra of this molecule [35].
So, a detailed interpretation of the vibrational spectra of the title compound has been made, in continuation of our theoretical studies, in the present work, we checked the relative performance of B3LYP, B3PW91, mPW1PW91 and PBEPBE methods at the SDD level taking the title compound as a test compound.
Experimental
In this work, we have found CeCl3 • 7 H2O as an efficient catalyst for the synthesis of the imine complexed with CeCl3 noted (P2-CeCl3) at room temperature through a one-pot reaction of aromatic aldehydes, and aromatic amines in methanol. It is also noteworthy to mention that our environmentally benign reaction does not generate any toxic waste products. A possible mechanism for the formation of the complex proposed by Mazaahir Kidwai et al[21] is shown in Figure 1.
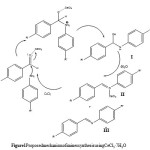 |
Figure 1: Proposed mechanism of imines synthesis using CeCl3 · 7 H2O Click here to View figure |
Instrumental Analysis
All reagents used for the synthesis of the compound are commercially available and were used without further purification. IR spectra were recorded on a Shimadzu FTIR-8400 S spectrophotometer as KBr pellets in the frequency range 4000 − 400 cm−1. 1H NMR and 13C NMR spectra were recorded on Nuclear magnetic resonance spectrometer type ”AVANCE 300MHz de BRUKER ” . Mass spectra (MS) were obtained by using electrospray ionization (ESI) technique.
Preparation of P2-CeCl3
Benzaldehyde (1 mmol) and para-methoxyaniline (1mmol) in MeOH (5 mL), were mixed and stirred at room temperature. To this, CeCl3 · 7 H2O (cerium chloride heptahydrate) (3 mol% ) was added. The progress of re- action mixture was monitored by TLC (using petroleum ether/AcOEt = 80:20 as an eluent). After completion of the reaction, the reaction mixture was evaporated under reduced pressure to give the powder product. The predicted structure of the product is shown in Figure 2, it was unambiguously established on the basis of their spectral analysis (IR, 1H, 13C NMR and MS mass spectral data).
Characterization
P2-CeCl3:(E)-N-benzylidene-4-methoxyaniline CeCl3
C14H13 NO-CeCl3: Yield: 96 % , RMN 1H (CDCl3) δppm: 8.47 (s, 1H, =CH); 7.90 − 7.23 (m, 5H, -C2H5);
6.95−6.59 (m, 5H, O–C2H5)); 3.82 (s, 3H, CH3). RMN13C (CDCl3) δppm: 158.42 (O-CH); 131.05 (=CH-C-CCH); 128.75 (=CH-C-CH); 122.22(O-C-C-CH); 114.77 (O-C-CH); 55.50 (CH3). SM: [M+] (m/z) = 457.73, found: 459.47.
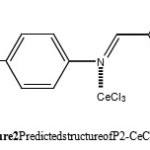 |
Figure 2: Predicted structure of P2-CeCl3 |
Computational details
In addition to the experimental study, we have used molecular orbital calculations to locate the most stable candidate structure, evaluate the accuracy of the geometries, provide the initial molecule model for structure solutions and investigate spectral properties of the studied complex. The DFT calculation of this com- pound (P2-CeCl3) were performed using GAUSSVIEW 5.08 molecular visualization program and the Gaussian 09 program package [33]. For calculations, involving the cerium (III), the Stuttgrat Dresden(SDD) effective core potential[34] basis set was used with the B3LYP, B3PW91, mPW1PW91 and PBEPBE methods, the vibrational frequencies were also calculated with these methods.
In the fitting processes performed to correct over estimations at the calculated harmonic frequencies, the values computed at these levels contain known systematic errors and therefore, scaling factors 0.9534, 0.9477, 0.9429 and 0.9795 for B3LYP, B3PW91, mPW1PW9,PBEPBE, respectively[34]. We have also calculated optimal scaling factors for all investigated methods. After this procedure, no imaginary mode was observed.
Results and Discussion
The prediction of vibrational wave numbers was done at DFT (B3LYP, B3PW91, mPW1PW91, PBEPBE) levels with SDD basis set, as it has been well recognized as an efficient theoretical chemistry tool for studying vibrational properties of variety of molecule, positive wave numbers for each normal mode confirm the stability of the molecular structure obtained with minimum energy. As already known, the wave numbers calculated are almost systematically over estimated compared to the experimentally measured values, these differences are widely corrected by scaling the calculated values with factors corresponding to the methods used. The complex study has 33 atoms, which possess 93 normal mode of vibration where the Gaussview 5.08 package has been considered to get visual animation and also for verification of these normal assignment vibrational modes (IR). The predicted infrared spectra of our com- pound are shown in Figure 3, a comparison between the calculated and the experimental data help us to understand the observed spectral frequencies, which are collected in two Tables 1 and 2. According with results in this Tables, experimental fundamentals are in better agreement with the scaled fundamentals and are found to have a good correlation for the four methods used, we also calculated the mean absolute deviation, root mean square, TS (tacking signal) and the mean absolute percentage error between the calculated and experimental data. Careful examining and comparison of the experimental and calculated values may conclude the following observations for each vibration mode:
 |
Figure 3: The theorethical frequencies (scaled); calculated with B3LYP, B3PW91, mPW1PW91, PBEPBE methods using SDD basis set Click here to View figure |
Imine group vibrations
The first type of vibration is the C=N stretching skeletal bands are observed in the range of 1650 −1500cm−1,[35–38] for the title compound, the strong ab- sorption at 1505 cm−1 due to νC=N frequencies. The calculated stretching vibration mode of the C=N (imine) group band of the complex with the four methods (see Figure 3 and Table 1 entry 18 for ν(C=N) at the SDD basis set are 1507, 1519, 1526 and 1531 cm−1 (scaled), respectively. The second , the deformation C-H ( see Table 1 entry 27) of the imine function , a band at 1363 cm−1, correspond to δC-H,(C=N), it is predicted at 1377, 1371, 1372 and 1385 cm−1, respectively[39].
Methoxy group
32 fundamentals vibrations can be associated to this group. Five stretching, seven deformation, one scis- soring, four umbrella, six rocking and nine torsional vibration modes are motion to the methyl group. The strongest band of CH3 asymmetric and symmetric stretching frequencies are established at 3026 ( 3037, 3035, 3038, 3055 cm−1), and 2879 (2884, 2879, 2882, 2895 cm−1) in infrared (the SDD calculations using the four methods) spectrum (see Figure 3 and entries 9 and 14 in Table 1). The band observed at 1442 cm−1 in the IR and 1442, 1445, 1450 and 1441 cm−1 (SDD calculation using the four methods) are assigned as the de- formation mode of CH3 (νCH3 entry 20 in Table 1), the scissoring vibration of this group appear at 1405
cm−1 (entry 24 in Table 1) and confirmed by the calculated frequencies in the same rang 1407, 1404, 1407 and
1409 cm−1 and 1391 cm−1 is attributed to the CH3 umbrella vibration (entry 25 in Table 1), the latter mode is well defined with the SDD calculation, give assignment similar to the experimental value. For the rocking vibrations, many bands observed but the clearest that can identify this vibration mode appears at 1028 cm−1 in IR
spectrum (entry 44 in Table 1), with values not close enough with SDD calculation at 984, 976, 980, and 980
cm−1. In the vibration spectrum of this compound four bands observed which could be assigned to the following modes with different internals coordinates: 1266 and 1207 cm−1 for νC-O vibrations, δO-C at 552 cm−1 and torsion vibration at 444 and 410 cm−1, observed in the same range with the calculated vibration.
Aromatic rings
The characteristic νC-H stretching vibrations of the aromatic ring are expected to appear in 3000-3100 cm−1
Frequency range[40,41]. Although nine vibrational bands are predicted in this rang, the νC-H stretching vibration of the title compound is assigned to two strong bands. The first corresponds to the ring para-substituted and
the second for the benzene ring at 3093 and 3067 cm−1, in IR, respectively (entry 1 and 2 in Table 1).
For the phenyl ring (ring2), the bands observed at 1207 cm−1 in IR spectrum is assigned as C-H deformation. Also SDD calculation give these mode at 1197, 1199, 1202 and 1211 cm−1, respectively, using the four methods (entry 33 Table 1). They are in agreement with the experimental band. On the other hand, the δC-H of the para substituted (ring2) are seen in the rang[35] 995-1315 cm−1;[42,43] band observed at 1266 cm−1 observed in the IR spectrum attributed to δC-H of ring2, for this mode the frequencies calculated are 1266, 1261,1263, and 1265 cm−1, respectively, with the four methods (entry 31 Table 1). In general, the C=C stretching vibration in aromatic compound are seen in the region of 1650-1430 cm−1.[32,44–46] A characteristic band appear at 1437 cm-1 confirmed by the SDD calculation (1436, 1435, 1439 and 1435 cm−1) attributed to νC-C frequency (entry 21 Table 1). For the modes of deformation in the plan of δC-C-C are observed at 820, 812,778, and 536 cm−1, confirmed by the calculated assignments.
Ce-N and Ce-Cl vibrations modes
There is not far experimental data for this type of complexes, the complexes with Ce(III) ligand bonding is strongly ionic with small donor-acceptor character and supposes weak and not informative bands[47], The Ce-N stretching vibrations are often coupled with other vibrational modes of the ring. For this purpose, the SDD calculation with the four methods offer explicit information and details about these modes vibrations, or you notice the appearance of this mode below 400 cm−1; the νCe-Cl is closest vibration, it appear at (356, 339, 343, and 336 cm−1) (entries 72, 74 and 75 Table 1), whereas the νCe-N stretching and δCe-Cl bending vibration appear in the same band at (145, 148, 150 and 142 cm−1).
Obviously, B3LYP functional give results in closest agreement with the experimental frequency over the other functional and for performance appraisal of the used methods, the Mean Absolute Deviation (MAD), Mean Absolute Percentage Error (MAPE) and Mean Square Values (RMS) were calculated and given in Table2. RMS values of the experimental and calculated IR bands are 16.64, 16.64, 17.45, 17.66 and MAPE values are 1.33, 1.39, 1.40, and 1.5 for B3LYP, B3PW91, mPW1PW91, PBEPBE methods respectively.
These results indicate a good agreement between theoretical and experimental data of the title compound at SDD basis set using B3LYP, B3PW91, mPW1PW91, PBEPBE functional and specially, B3LYP, which have the best agreement with the smallest difference between calculated and experimental frequencies.
 |
Table 1: Vibrational wave numbers obtained for the title compound at SDD basis set with B3LYP, B3PW91, mPW1PW91 , PBEPBE methods Click here to View table |
Finally, we proposed and calculated the optimal scaling factors, which are recommended to get a very close frequencies. The obtained scaling factors are 0.9439, 0.9382, 0.9335 and 0.9697 for B3LYP, B3PW91,
mPW1PW91, PBEPBE, respectively at the SDD basis set.
Molecular geometry
The molecular structure of the compound in the ground state (in vacuo) was optimized using density functional theory, The optimized geometrical parameters: bond length in Å, bond angles and dihedral angles in degree, determined with mPW1PW91, B3LYP, B3PW91 and PBEPBE methods at SDD basis set for molecule are collected in Table 2 in accordance with the atom numbering shown in Fig. 4. The values of the distances be- tween the atoms and valence or torsion angles are very close in any calculation method.
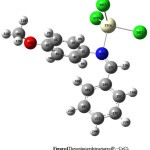 |
Figure 4: The optimized structure of P2−CeCl3 |
 |
Table 2: The selected bond lengths , valence angles and dihedral angles of the compound at SDD basis set with B3LYP, B3PW91, mPW1PW91 ,PBEPBE methods. Click here to View table |
Mulliken Atomic Charges
Mulliken atomic charge calculation in Table 3 has an important role in the application of quantum chemical calculation to molecular system because of atomic charges present a vital insight in the understanding of several kinds of chemical reactions and they are crucial to the elucidations of a number of other phenomena, effect dipole moment, molecular polarizability, electronic structure and more a lot of properties of molecular systems. In addition to the charge distribution on the molecule has an important influence on the vibrational spectra, they also are important factors in molecular structure-activity and structure-property relations.[46,48] The total atomic charges of the imine complexed with CeCl3 obtained by the Mulliken Population Analysis (MPA), they were calculated using B3LYP, B3PW91, PBEPBE and mPW1PW91 levels with SDD basis set, were listed in Table 3.
Our interest here is the comparison of the performance and assess the sensitivity of the calculated charges using different methods in order to describe electronic distribution in our complex and the influence of the presence of CeCl3, by comparing the MPA charges in the imine(P2) with imine complexed (P2-CeCl3).
In P2 and P2-CeCl3 compounds, more charge density was found at C14 linked to imine function (C=N) than that the other carbon atoms. The high positive charges at C6 is due to effect of oxygen atom (O26) of the methoxy group attached in the para position of ring 1 with C6. The charge distribution of the title compound shows that the carbon atoms attached with hydrogen atoms (C1, C2, C4, C5, C12, C15,C16,C17, C19, C21) are negative whereas the remaining carbon atoms are positively charged. The oxygen and nitrogen atoms have more negative charges whereas all hydrogen atoms have a positive charge. It is very clear from Table 3 that there is large change in Mulliken charges of the nitrogen atom (N11) in P2 and P2-CeCl3, and this variation reveals that there is charge transfer through the N-Ce bond, explained by the appearance of a strongly negative charge noted for the nitrogen atom, which is involved, as acceptor and the bigger positive charge is noticed on the cerium atom, as donor. This donor effect is due to the presence of electronegative nitrogen atoms; while the Cl atoms have a the highest negative charges in P2-CeCl3. All used methods have shown similar charges for each compound (imine and complex), where a change in charges distribution have been observed, for which the complex have the highest charges.
Table 3: MPA charges in imine (P2) and imine complexed (P2-CeCl3) at SDD basis set with B3LYP, B3PW91, mPW1PW91, PBEPBE methods
| Atom | B3LYP/SDD | B3PW91/SDD | PBEPBE/SDD | mPW1PW91/SDD | ||||
| P2-Cl3 | P2 | P2-Cl3 | P2 | P2-Cl3 | P2 | P2-Cl3 | P2 | |
| H31 | 0.227 | 0.205 | 0.237 | 0.215 | 0.244 | 0.222 | 0.239 | 0.218 |
| H32 | 0.265 | 0.231 | 0.271 | 0.237 | 0.279 | 0.245 | 0.275 | 0.241 |
| H33 | 0.224 | 0.203 | 0.234 | 0.213 | 0.242 | 0.219 | 0.235 | 0.215 |
| C30 | -0.47 | -0.474 | -0.497 | -0.449 | -0.529 | -0.536 | -0.504 | -0.506 |
| O29 | -0.275 | -0.313 | -0.290 | -0.327 | -0.225 | -0.268 | -0.303 | -0.338 |
| C1 | -0.339 | -0.323 | -0.356 | -0.327 | -0.348 | -0.333 | -0.360 | -0.344 |
| C2 | -0.291 | -0.364 | -0.281 | -0.367 | -0.314 | -0.381 | -0.275 | -0.372 |
| C3 | 0.223 | 0.220 | 0.193 | 0.227 | 0.207 | 0.211 | 0.181 | 0.229 |
| C4 | -0.258 | -0.303 | -0.249 | -0.304 | -0.225 | -0.317 | -0.257 | -0.308 |
| C5 | -0.311 | -0.333 | -0.329 | -0.342 | -0.324 | -0.349 | -0.343 | -0.349 |
| C6 | 0.360 | 0.327 | 0.395 | 0.360 | 0.334 | 0.311 | 0.407 | 0.371 |
| H7 | 0.269 | 0.231 | 0.273 | 0.237 | 0.281 | 0.243 | 0.278 | 0.242 |
| H8 | 0.275 | 0.242 | 0.279 | 0.241 | 0.292 | 0.252 | 0.282 | 0.247 |
| H9 | 0.269 | 0.250 | 0.274 | 0.252 | 0.285 | 0.262 | 0.286 | 0.258 |
| H10 | 0.283 | 0.245 | 0.284 | 0.246 | 0.291 | 0.254 | 0.292 | 0.252 |
| N11 | -0.341 | -0.034 | -0.371 | -0.067 | -0.288 | -0.017 | -0.387 | -0.077 |
| Ce25 | 0.562 | 0.576 | 0.495 | 0.571 | ||||
| Cl26 | -0.056 | -0.062 | -0.049 | -0.048 | ||||
| Cl27 | -0.043 | -0.040 | -0.037 | -0.035 | ||||
| Cl28 | -0.054 | -0.051 | -0.044 | -0.045 | ||||
| C12 | -0.251 | -0.434 | -0.213 | -0.400 | -0.277 | -0.461 | -0.202 | -0.396 |
| H13 | 0.267 | 0.208 | 0.265 | 0.210 | 0.273 | 0.220 | 0.270 | 0.215 |
| C14 | 0.400 | 0.395 | 0.399 | 0.385 | 0.414 | 0.413 | 0.394 | 0.377 |
| C15 | -0.296 | -0.354 | -0.286 | -0.345 | -0.311 | -0.363 | -0.289 | -0.350 |
| C16 | -0.280 | -0.311 | -0.275 | -0.302 | -0.298 | -0.321 | -0.275 | -0.303 |
| C17 | -0.221 | -0.223 | -0.234 | -0.231 | -0.230 | -0.237 | -0.242 | -0.237 |
| C19 | -0.233 | -0.231 | -0.246 | -0.239 | -0.240 | -0.245 | -0.256 | -0.246 |
| C21 | -0.177 | -0.221 | -0.185 | -0.227 | -0.196 | -0.236 | -0.191 | -0.232 |
| H18 | 0.244 | 0.224 | 0.243 | 0.224 | 0.255 | 0.243 | 0.248 | 0.229 |
| H20 | 0.260 | 0.262 | 0.262 | 0.265 | 0.271 | 0.274 | 0.266 | 0.270 |
| H22 | 0.259 | 0.223 | 0.260 | 0.224 | 0.269 | 0.234 | 0.266 | 0.230 |
| H23 | 0.252 | 0.225 | 0.251 | 0.225 | 0.262 | 0.236 | 0.257 | 0.231 |
| H24 | 0.259 | 0.225 | 0.260 | 0.226 | 0.262 | 0.236 | 0.266 | 0.232 |
Thermodynamic Properties
The thermodynamic data can be used to compute the other thermodynamic energies according to relation- ships of thermodynamic functions and estimate directions of chemical reactivity according to the second law of thermodynamics in thermo chemical field, from which the relations among energy, structural and reactivity characteristics of the molecules can be clarified. Knowledge of permanent dipole moment of a molecule helps to determine the molecule’s conformations.[49]. On the basis of vibrational analysis and statistical thermodynamics. The thermodynamic parameters for the title compound were obtained from the theoretical harmonic frequencies namely heat capacity, entropy, rotational constants, vibration and vibrational zero point energies of the compound have also been computed at the DFT-B3LYP, B3PW91, mPW1PW91 and PBEPBE levels using SDD basis set are presented in Table 4.
To better understand the complex formation, we note that there is a very large differences between the total energies and dipole moments of imine and the complex with used methods, this great difference shows the low reactivity or the high stability behavior, this explains the complex formation further than the imine formation. On un other hand, the high value of dipole moment of the P2-CeCl3 relative to that of the P2, could partly explain the high stability of the complex, which it is due to the presence of the donor effect of cerium which makes the nitrogen atom strongly electronegative. The biggest value of zero-point vibrational energy (ZPVE) is 153.56 kJ/mol obtained with mPW1PW91 whereas the smallest one is 147.72 kJ/mol obtained with PBEPBE, While the total energies calculated by the four levels are very close.
Table 4: Theoretical computed Energy(a.u), Zero Energy, (Kcal/mol), Rotational Constant(GHz), Entropy (cal/mol.K) and Dipole moment D(Kcal/mol.K)
| B3LYP/SDD | B3PW91/SDD | PBEPBE/SDD | mPW1PW91/SDD | |||||
| Thermodynamic Pa- rameters | P2-Cl3 | P2 | P2-Cl3 | P2 | P2-Cl3 | P2 | P2-Cl3 | P2 |
| Total En-ergy(Thermal): Etot (kcal/mol) | 165.231 | 156.944 | 166.001 | 157.667 | 161.658 | 153.329 | 167.057 | 158.735 |
| Heat capacity at const. vol.: Cv(cal/mol.K) | 74.507 | 52.474 | 74.253 | 52.296 | 76.233 | 54.144 | 73.769 | 51.870 |
| Entropy:S(cal/mol) | 169.201 | 119.397 | 160.677 | 119.322 | 170.727 | 120.427 | 167.159 | 119.007 |
| Vibrational energy: Evib(kcal/mol) | 163.453 | 155.167 | 164.223 | 155.889 | 159.881 | 151.552 | 165.280 | 156.958 |
| Zero-point vibrational energy:Eφ(kcal/mol) | 151.57885 | 148.44611 | 152.39319 | 149.18289 | 147.72441 | 144.61031 | 153.56278 | 150.30798 |
| Rotational con- stants(Ghz) | ||||||||
| X | 0.20233 | 1.23352 | 0.20685 | 1.22372 | 0.20395 | 1.24573 | 0.20867 | 1.21809 |
| Y | 0.15980 | 0.24847 | 0.16106 | 0.25261 | 0.15923 | 0.24380 | 0.16221 | 0.25575 |
| Z | 0.10552 | 0.22214 | 0.10717 | 0.22559 | 0.10556 | 0.21826 | 0.10818 | 0.27792 |
| Dipole moment (De- bye) | ||||||||
| µX | -8.7912 | -0.4915 | -8.6382 | -0.5134 | -7.1017 | -0.2950 | -8.5772 | -0.5151 |
| µY | 3.6801 | -1.6588 | 4.2952 | -1.7136 | 5.8090 | -1.6631 | 4.1951 | -1.7213 |
| µZ | 1.4217 | -1.3257 | 1.4468 | -1.3238 | 1.5532 | -1.2643 | -1.3979 | -1.3253 |
| µtotal | 9.6359 | 2.1796 | 9.7550 | 2.2254 | 9.3055 | 2.1098 | 9.6500 | 2.2326 |
| Total energy (a.u) | -2526.910432 | -671.1557335 | -2526.546118 | -670.901587 | -2525.447260 | -670.355180 | -2526.792928 | -670.9971721 |
Conclusion
The comprehensive studies of the molecular structure, Mullikan atomic charges and thermodynamic parameters of the complex formed (E)-N-benzylidene-4- methoxyaniline, CeCl3 suggested that the ligand acts as tetradentate one in the Ce(III) complex formation which is favored over the formation of imine, confirming the utility of the this catalyst (CeCl3) in the synthesis of imines and the nitrogen derivatives or imines training in situ. The comparison of the experimental and calculated frequencie , show that the experimental values are in a good agreement with SDD calculation results and obviously B3LYP function which has given the closets results. Optimal uniform factors were also calculated. Taking into account small variations between calculated and experimental data, we can recommend new scaling factors of 0.9439, 0.9382, 0.9353 and 0.9697 for B3LYP, B3PW91, mPW1PW91 and PBEPBE levels, respectively, at SDD basis set, for the IR spectral future predictions for unknown compounds of this class.
References
- Rezaeivala, M.; Keypour, H. Coordination Chemistry Reviews 2014, 280, 203.
CrossRef - Hayashi, T.; Shibata, H.; Orita, S.; Akitsu, T. European Chemical Bulletin 2012, 2 49.
- Awual, M. R.; Hasan, M. M.; Shahat, A.; Naushad, M.; waku, H. S.-.; Yaita, T. Chemical Engineering Journal 2015, 265, 210.
CrossRef - Terzis, A.; Mentzafos, D.; Tajmir-Riahi, H. Inorganica Chimica Acta 1984, 84, 187.
CrossRef - Afkhami, A.; Madrakian, T.; Shirzadmehr, A.; Tabatabaee, M.; Bagheri, H. Sensors and Actuators B: Chemical 2012, 174 237
- Afshar, S.; Bullock, J. I. Inorganica Chimica Acta 1980, 38, 145.
CrossRef - Fenton, D. E.; Vigato, P. A. Chem. Soc. Rev 1988, 17, 69.
CrossRef - A.Mouadili; I.Lakehal; A.Takfaoui; F.Halaimia; H.Nacer; M.L.Hamlaoui; B.Hammouti; M.Messali; R.Touzani J. Mater. Environ. Sci. 2014, 5, 715.
- Takfaoui, A.; Lakehal, I.; Bouabdallah, I.; Halaimia, F.; Nacer, H.; Hammouti, B.; Touzani, R. J. Mater. Environ. Sci. 2014, 5, 753.
- Piro, N. A.; Robinson, J. R.; Walsh, P. J.; Schelter, E. J. Coordination Chemistry Reviews 2014, 260, 21.
CrossRef - Zamani, H. A.; Ganjali, M. R.; Adib, M. Sensors and Actuators B: Chemical 2007, 120, 545.
CrossRef - Maity, D.; Chattopadhyay, S.; Ghosh, A.; Drew, M. G.; Mukhopadhyay, G. Inorganica Chimica Acta 2011, 365, 25.
CrossRef - Kocyigit, O.; Kursunlu, A. N.; Guler, E. Journal of hazardous materials 2010, 183, 334.
CrossRef - Bhowmik, P.; Drew, M. G.; Chattopadhyay, S. Inorganica Chimica Acta 2011, 366, 62.
CrossRef - Esmaielzadeh, S.; Azimian, L.; Shekoohi, K.; Esfandiari, H.; Asadi, M.; Zare, Z.; Nejad, A. R.; Mohammadi, K. Inorganica Chimica Acta 2013, 405, 155.
CrossRef - Ali, M. A.; Haroon, C. M.; Nazimuddin, M.; jumder, S. M.-u.-H. M.-.; Tarafder, M. T.; Khair, M. A. Transition Metal Chemistry 1992, 17, 133.
CrossRef - Hossain, M. E.; Alam, M. N.; Ali, M. A.; Nazimuddin, M.; Smith, F. E.; Hynes, R. C. Polyhedron 1996, 15, 973.
CrossRef - Tan, X.-J.; Liu, H.-Z.; Ye, C.-Z.; Lou, J.-F.; Liu, Y.; Xing, D.-X.; Li, S.-P.; Liu, S.-L.; Song, L.-Z. Polyhedron 2014, 71, 119.
CrossRef - Bagihalli, G. B.; Avaji, P. G.; Patil, S. A.; Badami, P. S. European journal of medicinal chemistry 2008, 43, 2639.
CrossRef - Sathisha, M.; Shetti, U. N.; Revankar, V.; Pai, K. European journal of medicinal chemistry 2008, 43, 2338.
CrossRef - RAIKWAR, K.; AGARWAL, D. D. Orient. J. Chem., 2015, 31, 547.
- ABDULFATAI, A. S.; ADAMU, U.; SULAIMAN, I.; HAMZA2, A. Orient. J. Chem., Vol. 31(4), (2015) 2015, 31, 1985.
- AL-KHODIR, F. A. I. Orient. J. Chem 2015, 31, 1277.
CrossRef - Fan, Y.-H.; Wang, A.-D.; Bi, C.-F.; Xiao, Y.; Bi, S.-Y.; Zhang, X.; Wang, Q. Synthetic Metals 2011, 161, 1552.
CrossRef - Silva, C. M. d.; Silva, D. L. d.; Modolo, L. V.; Alves, R. B.; Resende, M. A. d.; Martins, C. V.; Fa´tima, A. d. Journal of Advanced research 2011, 2, 1.
CrossRef - Kidwai, M.; Anwar, J. Journal of the Brazilian Chemical Society 2010, 21, 2175.
CrossRef - Sindhu, Y.; Athira, C.; Sujamol, M.; Joseyphus, R. S.; hanan, K. M.-. Metal-Organic, and Nano-Metal Chemistry 2013, 43, 226.
CrossRef - Wang, Q.; Huang, Y.; Zhang, J.-S.; Yang, X.-B. Bioinorganic Chemistry and Applications 2014, 2014, 9.
- Chinthaparthi, R. R.; Gangireddy, C. S. R.; Mudumala, V. R.; Gundluru, M.; Chamarthi, N.; Cirandur, S. R. Tetrahedron Letters 2013, 54, 6071.
CrossRef - Ubani, O. C.; Ngochindo, R. I.; Odokuma, L. O. J. Chem. Pharm. Res 2015, 7, 720.
- Adly, O. M.; Taha, A.; Fahmy, S. A. Journal of Molecular Structure 2015, 1093, 228.
CrossRef - El-Wahab, Z. A.; Mashaly, M.; Faheim, A. Chem Pap 2005, 59, 25.
- Shebl, M. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2008, 70, 850
CrossRef - Montazerozohori, M.; Tavakol, H.; Kheiri, S.; Musavi, S.; Joohari, S. Bulgarian chemical communications 2014, 46, 96.
- Glendening, E. D.; Feller, D.; Thompson, M. A. Journal of the American Chemical Society 1994, 116, 10657.
CrossRef - Gabal, M.; Elroby, S. A.; Obaid, A. Powder Technology 2012, 229, 112.
CrossRef - Yabalak, E.; Gu¨nay, F.; Kasumov, V. T.; Arslan, H. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2013, 110, 291.
CrossRef - Frisch, M.; Trucks, G.; Schlegel, H. B.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; son, G. P.-. Gaussian 09, revision a. 02, gaussian, Inc., Walling- ford, CT 200.
- Yang, Y.; Gao, H. Journal of Molecular Structure 2012, 1013, 111.
CrossRef - Anto, P.; Panicker, C. Y.; Varghese, H. T.; Philip, D.; Arpaci, O. T.-.; Tekiner-Gulbas, B.; Yildiz, I. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2007, 67, 744.
CrossRef - Saxena, R.; Kandpal, L.; Mathur, G. Journal of Polymer Science Part A: Polymer Chemistry 2002, 40, 3959.
CrossRef - Bellamy, L. J. Chapman and Hall London 1975, 1.
- Xie, J.; Chang, J.; Wang, X. Application of ir in organic chemistry and medicinal chemistry 2001.
- Li, L.; Cai, T.; Wang, Z.; Zhou, Z.; Geng, Y.; Sun, T. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2014, 120, 106.
CrossRef - Mary, Y. S.; Jojo, P.; Panicker, C. Y.; Alsenoy, C. V.; Ataei, S.; Yildiz, I. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2014, 122, 499.
CrossRef - Roeges, N. P. Wiley 1994.
- Kumar, V.; Panikar, Y.; Palafox, M.; Vats, J.; Kostova, I.; Lang, K.; Rastogi, V. Indian Journal of Pure & Ap- plied Physics 2010, 48, 85.
- Kalsi, P. New Age Inter- national 2007.
- Sathyanarayana, D. N. New Age International 2007.
- Kostova, I.; Momekov, G. European journal of medicinal chemistry 2008, 43, 178.
CrossRef - Sinha, L.; Prasad, O.; Karabacak, M.; Mishra, H.; Narayan, V.; Asiri, A. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2014, 120, 126.
CrossRef - Arjunan, V.; Govindaraja, S. T.; Ravindran, P.; Mohan, S. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2014, 120, 473.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


