Synthesis and Antihypertensive Activity of Some Novel Pyridazinones
Mohd. Imran* and Naira Nayeem
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Northern Border University, Rafha – 91911, P.O. BOX 840, Kingdom of Saudi Arabia. Corresponding Author Email: imran_inderlok@yahoo.co.in
DOI : http://dx.doi.org/10.13005/ojc/320129
Article Received on :
Article Accepted on :
Article Published : 03 Mar 2016
Four pyridazinones (2-5) were synthesized by the reaction of 3-benzoylpropionic acid with the corresponding hydrazines. These pyridazinones were further derivatized with appropriate aromatic aldehydes in the presence of piperidine to obtain the desired compounds (6-13). The chemical structures of the synthesized pyridazinones were elucidated on the basis of their spectral analysis (IR, 1H-NMR, and 13C-NMR). Molecular docking studies were carried out to identify the most active antihypertensive pyridazinone compound by using the crystal structure of human Angiotensin Converting Enzyme (ACE), the enzyme implicated in the pathogenesis of hypertension. The compound (6) was identified as the most active inhibitor of the Angiotensin Converting Enzyme (ACE). This compound was further assessed for its ACE inhibitory activity using Dojindo ACE Kit-WST test kit. The enzymatic ACE inhibitory activity revealed that the compound (6) had IC50 value of 5.78 μg/mL, wherein the standard drug Lisinopril had IC50 value of 0.85 μg/mL. It has been concluded that incorporation of free amino groups and free carboxylic acid groups in the structure of the synthesized pyridazinone (6-13) may provide more active and potent ACE inhibitors.
KEYWORDS:Benzoylpropionic acid; Pyridazinones; Molecular modelling; ACE inhibitor; Antihypertyensive agents
Download this article as:| Copy the following to cite this article: Imran M, Nayeem N. Synthesis and Antihypertensive Activity of Some Novel Pyridazinones. Orient J Chem 2016;32(1). |
| Copy the following to cite this URL: Imran M, Nayeem N. Synthesis and Antihypertensive Activity of Some Novel Pyridazinones. Orient J Chem 2016;32(1). Available from: http://www.orientjchem.org/?p=14540 |
Introduction
Cardiovascular disease is a major public health problem worldwide and also accounts for approximately 30% of all deaths1. It is also considered as the major cause of death in the Kingdom of Saudi Arabia2-4.According to the literature2-4, the burden of hypertension remained very high in KSA from 1990 to 2010; the number of deaths resulting from ischemic heart disease and hypertensive heart disease in the Middle East and North Africa region (including the GCC countries) is estimated to be 294/100,000 and 115/100,000 respectively; and the number of disability-adjusted life years (DALYs) resulting from ischemic and hypertensive heart disease is 3702/100,000 and 1389/100,000, respectively, in the same region. These studies further state that KSA has a young population with 81% of the population under the age of 40 and life expectancy has increased from 72.5 and 76.3 years in 1990 to 75.0 and 79.9 years in 2010 for men and women, respectively. As life expectancy increases and the size of population is on the rise, the burden of hypertension and other chronic diseases, if uncontrolled, will pose major challenges to the health system. Studies have also revealed that there is a need for more research in the field of cardiovascular disease in developing countries because in these countries there would be an increase in the prevalence of cardiovascular disease in all age group of patients5,6. There are many cardioactive pyridazinone derivatives, which are either in clinical use or have been tested in clinical trials, for example, imazodan7,8, CI-9308,9, pimobendan8,10, indolidan8,11, levosimendan8,12, SK&F-937418,13, Y-5908,14, Meribendan15, NSP-80416, NSP-80516, bemoradan17,18, senazodan19, amipizone8,18, prinoxodan20, SKF 9565421, siguazodan22 and KF 1523222. Some of these pyridazinone derivatives were found not suitable for further development because of their side effects and low potency. Keeping this in mind, it was thought to synthesize some novel pyridazinone derivatives as antihypertensive agents that may be suitable for further development.
Materials and Methods Chemistry
Melting points were measured in open capillary tubes and are uncorrected. IR (KBr) spectra were recorded on a JASCO, FTIR-4100 spectrophotometer. The 1H-NMR and 13C-NMR spectra were recorded on Bruker Ultrashield 500 Plus MHz spectrophotometer. All reagents used in the present work were of analytical grade. The standard drug Lisinopril used for the assessment of in vitro ACE inhibitory activity was procured from Sigma Aldrich, USA. Purity of the compounds was checked on silica gel G plates using iodine vapours as visualizing agent. Rf value of the compounds was determined by using a mixture of benzene and acetone (9:1). The synthetic pathway for the preparation of the title pyridazinones(2-13) is provided in Figure 1.
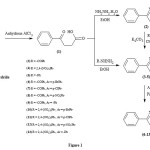 |
Figure 1 Click here to View figure |
Synthesis of 3-Benzoylpropionic acid (1)
To a solution of succinic anhydride (0.1 mole) in benzene (50 mL), anhydrous aluminium chloride(0.125 mole) was added in small portions with stirring over a period of 2 hours. The reaction mixture was refluxed for 2 hours. After completion of the reaction, excess of benzene was removed by steam distillation. The reaction mixture was dissolved in sodium hydroxide solution, filtered, and hydrochloric acid was added to it. The solid mass obtained was filtered, washed with cold water, dried and recrystallized from methanol to give a colorless product which gave effervescence with sodium bicarbonate solution23-25.
Synthesis of 6-phenyl-4,5-dihydropyridazin-3(2H)-one (2)
A mixture of 3-benzoylpropionic acid (0.01 mole) and hydrazine hydrate (0.015 mole) in ethanol (25 mL) was refluxed for about 6 hours. The resulting mixture was cooled at room temperature. The solid obtained was filtered, washed with dilute solution of sodium bicarbonate and then recrystallized from ethanol.
Synthesis of 2-benzoyl-6-phenyl-4,5-dihydropyridazin-3(2H)-one (3)
6-Phenyl-4,5-dihydropyridazin-3(2H)-one (2) (0.01 mole) was taken in a 100 mL round bottom flask. Potassium carbonate (0.015 mole) and acetone (25 mL) were added to the flask. The mixture was stirred at room temperature and benzoyl chloride (0.01 mole) was added drop wise in 20 to 30 minutes. The resulting mixture was stirred at room temperature for about 15 hours. The resulting mixture was reduced to half of its volume and then poured to crushed ice. The solid separated was washed with water, dilute sodium bicarbonate solution and recrystallized from ethanol. Alternatively, this compound may also be prepared by the reaction of benzoic acid hydrazide and 3-benzoylpropionic acid.
Synthesis of 2-(2,4-dinitrophenyl)-6-phenyl-4,5-dihydropyridazin-3(2H)-one (4)
A mixture of 3-benzoylpropionic acid (0.01 mole) and 2,4-dinitrophenyl hydrazine (0.01 mole) in ethanol (30 mL) was refluxed for about 10 hours. The solid obtained was hot filtered, washed with dilute solution of sodium bicarbonate and then recrystallized from ethanol.
Synthesis of 2,6-diphenyl-4,5-dihydropyridazin-3(2H)-one (5)
The compound (5) was also prepared by same method used for the compound (4), wherein 2,4-dinitrophenyl hydrazine was replaced by phenyl hydrazine.
Synthesis of 2-benzoyl-4-(4-bromobenzylidene)-6-phenyl-4,5-dihydropyridazin-3(2H)-one (6)
A mixture of 2-benzoyl-6-phenyl-4,5-dihydropyridazin-3(2H)-one (4) (0.01 mole) and p-bromo benzaldehyde (0.01 mole) was taken in a round bottom flask containing 25 mL of absolute ethanol. Piperidine (0.5 mL) was added to the mixture and it was refluxed for about 8 hours. The mixture was reduced to half of its volume and then poured to crushed ice. The solid separated was washed with water, and recrystallized from ethanol. The compounds (7-13) were also prepared by similar methods.
Molecular Modelling Studies
The compounds (6-13) were subjected for their molecular modelling docking studies. All computations were performed on an Intel(R) Core(TM) i5-2400 CPU @ 3.10 GHz capacity processor with memory of 8 GB RAM running with the Window 7 operating system. Glide 5.9 implemented in the Maestro 9.4 software package (Schrodinger, LLC, New York, NY, 2013) was used for docking studies. Angiotensin Converting Enzyme (ACE) is an enzyme that is involved in the pathogenesis of hypertension26. The crystal structure of human Angiotensin Converting Enzyme (ACE) protein (PDB code: 1O86, Resolution – 2.00 Å) was retrieved from Protein Data Bank (PDB) and was utilized for molecular modelling studies27. Protein was prepared with the Protein Preparation Wizard in Maestro using options: bond orders were assigned, hydrogen atoms were added, formal charges were treated and water molecules were deleted. Hydrogen bonding network was then optimized using the exhaustive sampling option and the protein was minimized to an RMSD limit from the starting structure of 0.3 Å using the Impref module of Impact with the OPLS_2005 force field. Prepared protein structure was used to generate Glide scoring grids for the subsequent docking calculations. Docking grids were generated with the default settings in Glide using the co-crystalized ligand to define the center of the grid box (20×20×20 Å). Default parameters were used and no constraints were included during grid generation. The three dimensional coordinates of most active synthesized compound (6) and lisinopril was generated using Maestro module of Schrodinger. Ligands were prepared using LigPrep 2.6 with Epik 2.4 to expand protonation and tautomeric states at 7.0 ± 2.0 pH units and energy was minimized using the OPLS 2005 force field. The energy minimized ligands were docked to the protein structure using Glide XP docking since it incorporates structural and energetic information using the scoring function28.
Angiotensin Converting Enzyme Inhibitory Assay
The compound (6) that showed highest ACE inhibitory activity according to the molecular modelling studies was subjected to ACE inhibitory assay using Dojindo ACE Kit-WST test kit, Dojindo Laboratories, Kumamoto, Japan29,30. Briefly, The enzymatic reaction was initiated by the ACE and aminoacylase in the mixture containing 3HB-GGG (3-hydroxybutyrate glycylglycylglycine) and the ACE-inhibitor. The mixture was then incubated at 37oC for 60 minutes. During this incubation, the substrate, 3HB-GGG, was enzymatically cut into 3HB-G and G-G and then 3HB and G. The yield of 3HB was monitored indirectly through formazan concentration, which was measured at 450 nm after 10 minute reaction at 25oC. Testing procedures were run according to the manufacturer’s instructions using a 96-well plate without modification, and the inhibition rate was calculated based on a comparison of the optical absorbance of samples-treated wells As, control wells AC, and blank wells Ab. Absorbance was measured at 450 nm using the microplate reader Biotek-ELX800 (BioTek, Vermont, USA). Inhibition rates were calculated using the following equation.
Inhibition rate (%) = [Ac-As/Ac-Ab]x100
Statistical analysis
All ACE inhibitory activity data are presented as mean ± standard deviation (SD, n = 3). The data were analyzed by one-way analysis of variance (ANOVA) with Dunnett’s Multiple Comparison Test with respect to control group and standard groups using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego California USA). The results were considered significantly different at p < 0.05. The IC50 values were determine by linear regression calculator of GraphPad software.
Results and Discussion
Chemistry
The compounds (1-13) were prepared according to the method outlined in Figure 1. The melting point of the compound (1) was in accordance with the literature23-25. The physical constants of the synthesized compounds (1-13) are summarized in Table 1.
Table 1: The physical constants of the synthesized compounds
|
Compd. No. |
R |
Ar |
Molecular Formula |
M.P. (±2oC) |
Yield (%) |
Rf Value |
|
1 |
– |
– |
C10H10O3 |
115 |
75 |
0.82 |
|
2 |
– |
– |
C10H10N2O |
152 |
80 |
0.77 |
|
3 |
-COPh |
– |
C17H14N2O2 |
166 |
55 |
0.76 |
|
4 |
2,4-(NO2)2Ph- |
– |
C16H12N4O5 |
188 |
80 |
0.63 |
|
5 |
-Ph |
– |
C16H14N2O |
144 |
60 |
0.77 |
|
6 |
-COPh |
p-BrPh- |
C24H17BrN2O2 |
175 |
60 |
0.74 |
|
7 |
-COPh |
p-ClPh- |
C24H17ClN2O2 |
155 |
65 |
0.76 |
|
8 |
-COPh |
p-NO2Ph- |
C24H17N3O4 |
175 |
50 |
0.78 |
|
9 |
-COPh |
-Ph |
C24H18N2O2 |
164 |
60 |
0.71 |
|
10 |
2,4-(NO2)2Ph- |
p-BrPh- |
C23H15BrN4O5 |
210 |
55 |
0.61 |
|
11 |
2,4-(NO2)2Ph- |
p-ClPh- |
C23H15ClN4O5 |
204 |
60 |
0.63 |
|
12 |
2,4-(NO2)2Ph- |
p-NO2Ph- |
C23H15N5O7 |
222 |
60 |
0.64 |
|
13 |
2,4-(NO2)2Ph- |
-Ph |
C23H16N4O5 |
196 |
70 |
0.63 |
The structures of the compounds (2-6) were confirmed on the basis of their IR, 1H-NMR, and 13C-NMR.The appearance of the IR absorption peaks from 1685 cm-1 to 1625 cm-1confirmed the stretching vibration of C=O group present at C-3 of the compounds (2-5). The appearance of the signals in the 1H-NMR spectra of the compounds (2-5) at δ (ppm) values from 2.36 to 2.44 confirmed two protons of -CO-CH2– and from 2.72 to 3.11 confirmed two protons of -C(=N)-CH2– portion of the pyridazinone ring. The compound (2) also showed characteristic signal at δ (ppm) value of 10.99 due to the –NH- proton. The structure of the compound (6) was supported by the appearnce of two stretching vibrations at 1685 cm-1 and 1645 due to two C=O groups. The absence of the peak for two protons of -CO-CH2– portion of the pyridazinone ring and appearnce of signals at δ (ppm) value of 2.88 due to the two protons of -C(=N)-CH2– portion of the pyridazinone ring confirmed the structure of the compound (6). The 13C-NMR of the compounds (2-6) were also consistent with their assigned structures. The detailed spectral data of the compounds (2-6) is provided below.
Phenyl-4,5-dihydropyridazin-3(2H)-one (2)
IR (KBr) cm-1: 3220 (N-H), 3100 (C-H), 1680 (C=O), 1610 (C=N), 1510 (C=C); 1H-NMR (DMSO-d6) δ ppm: 2.44 (t, J = 8 Hz, 2H, -CO-CH2-), 2.89 (t, J = 8 Hz, 2H, -C(=N)-CH2-), 7.40 (d, J= 11Hz, 3H, Ar-H), 7.74 (d, J= 11Hz, 2H, Ar-H), 10.99 (s, 1H, -NH-); 13C-NMR (DMSO-d6) δ ppm: 167.02 (C=O), 149.34, 135.90, 129.19, 128.42 (2C), 125.56, 125.31, 25.94, 21.85.
Benzoyl-6-phenyl-4,5-dihydropyridazin-3(2H)-one (3)
IR (KBr) cm-1: 1680 and 1640 (C=O), 1610 (C=N), 1505 (C=C); 1H-NMR (DMSO-d6) δ ppm: 2.44 (t, J = 8 Hz, 2H, -CO-CH2-), 2.90 (t, J = 8 Hz, 2H, -C(=N)-CH2-), 7.30-7.99 (m, 10H, Ar-H); 13C-NMR (DMSO-d6) δ ppm: 167.37 and 167.00 (C=O), 149.33, 135.89, 132.72 (2C), 129.24 (2C), 129.14, 128.43 (2C), 128.37, 125.53 (3C), 25.93, 21.85.
(2,4-Dinitrophenyl)-6-phenyl-4,5-dihydropyridazin-3(2H)-one (4)
IR (KBr) cm-1: 1625 (C=O), 1600 (C=N), 1510 (C=C), 1310 and 1340 (NO2); 1H-NMR (DMSO-d6) δ ppm: 2.36 (t, J = 8 Hz, 2H, -CO-CH2-), 3.02 (t, J = 8 Hz, 2H, -C(=N)-CH2-), 7.42-7.66 (m, 4H, Ar-H), 7.84-7.93 (m, 2H, Ar-H), 8.21-8.26 (m, 1H, Ar-H), 8.78 (s, 1H, Ar-H); 13C-NMR (DMSO-d6) δ ppm: 168.02 (C=O), 146.50, 144.40, 143.30, 136.09, 129.60, 128.51 (3C), 126.52 (2C), 123.43, 123.08, 116.45, 33.53, 23.63.
2,6-Diphenyl-4,5-dihydropyridazin-3(2H)-one (5)
IR (KBr) cm-1: 1685 (C=O), 1600 (C=N), 1510 (C=C); 1H-NMR (DMSO-d6) δ ppm: 2.72 (t, J = 8 Hz, 2H, -CO-CH2-), 3.11 (t, J = 8 Hz, 2H, -C(=N)-CH2-), 7.28 (t, J = 8Hz, 1H, Ar-H), 7.42-7.46 (m, 5H, Ar-H,), 7.57 (d, J = 11 Hz, 2H, Ar-H), 7.84 (t, J = 8 Hz, 2H, Ar-H); 13C-NMR (DMSO-d6) δ ppm: 165.25 (C=O), 151.84, 141.40, 135.40, 129.77, 128.54 (2C), 128.28 (2C), 126.11, 126.05 (2C), 124.84 (2C), 27.32, 22.23.
Benzoyl-4-(4-bromobenzylidene)-6-phenyl-4,5-dihydropyridazin-3(2H)-one (6)
IR (KBr) cm-1: 1685 and 1645 (C=O), 1605 (C=N), 1500 (C=C); 1H-NMR (DMSO-d6) δ ppm: 2.88 (s, 2H, -C(=N)-CH2-), 6.86-7.98 (m, 15H, Ar-H); 13C-NMR (DMSO-d6) δ ppm: 167.89 and 167.53 (C=O), 161.70, 149.87, 136.34, 136.01, 133.19 (2C), 131.21, 130.71, 129.70 (4C), 128.87 (4C), 126.01 (2C), 119.53, 117.49, 113.31, 26.39.
The compounds (7-13) were identified on the basis of the difference in the melting points and Rf values with respect to their reactants. These compounds were not subjected for their spectral analysis as their ACE inhibitory activity results .i.e. docking score obtained from the molecular modelling studies were not significant.
Molecular Modelling Studies
The molecular modelling studies revealed that almost all the functional groups of Lisinopril (docking score: -11.36; Figure 2) were occupied with either hydrogen bonding or other non-covalent lipophilic interactions and were important in exerting greater affinity towards ACE enzyme and ultimately ACE inhibition activity. On the other hand, the compound (6) (docking score: -6.14; Figure 3) was identified as the most active ACE inhibitor among the synthesized compounds. The compound(7) (docking score: -4.70), compound (8) (docking score: -4.78), compound (9) (docking score: -4.60), compound (10) (docking score: -5.02), compound (11) (docking score: -4.57), compound (12) (docking score: -4.13), and the compound (13) (docking score: -4.65) also showed some ACE inhibitory activity. The ACE inhibitory activity of the compounds (7-13) by molecular docking score was not found significant when compared to the standard drug Lisinopril.In general terms, larger is the docking score in negative value, more is the active binding with the target receptor .e.g. active site of ACE.
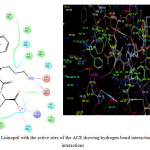 |
Figure 2: 2D/3D interaction of Lisinopril with the active sites of the ACE showing hydrogen bond interaction and non-covalent lipophilic interactions Click here to View figure |
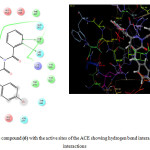 |
Figure 3: 2D/3D interaction of the compound (6) with the active sites of the ACE showing hydrogen bond interaction and non-covalent lipophilic interactions Click here to View figure |
Angiotensin Converting Enzyme Inhibitory Assay
Based on the molecular modelling results, the compound (6) was selected for further in vitro ACE inhibitory assay using Dojindo ACE Kit-WST test kit, Dojindo Laboratories, Kumamoto, Japan. The docking score and the ACE inhibitory activity data of the compound (6)with respect to the standard drug Lisinopril is provided in Table 2.
Table 2: In vitroACE inhibitory activity of Compound (6) and standard drug Lisinopril
|
Compound |
Concentration (μg/mL) |
%ACE Inhibition (Mean ± SD) |
IC50 (μg/mL) |
Docking Score |
|
Lisinopril |
1 |
43.20 ± 0.30* |
0.85 |
-11.36 |
|
2 |
60.30 ± 0.10* |
|||
|
4 |
75.63 ± 0.06* |
|||
|
8 |
86.66 ± 0.15* |
|||
|
Compound (6) |
1 |
12.50 ± 0.10* |
5.78 |
-6.14 |
|
2 |
28.80 ± 0.10* |
|||
|
4 |
49.70 ± 0.10* |
|||
|
8 |
58.76 ± 0.15* |
*p < 0.0001; n = 3.
The in vitroACE inhibitory activity revealed that the compound (6) had IC50 value of 5.78 μg/mL, wherein the standard drug Lisinopril had IC50 value of 0.85 μg/mL. These results were also supported by the results of molecular modelling, wherein it was evident that the compound (6) (Figure 3) did not bind effectively with the active site of the ACE enzyme as compared to standard drug Lisinopril (Figure 2). This may be due to the fact that the compound (6) does not have free functional groups that aid in the binding with the active site of the ACE enzyme. It is possible that the incorporation of free amino groups and free carboxylic acid groups in the structure of the compounds(6-13) may provide better and more potent ACE inhibitors.
Conclusion
Based on the molecular modelling results and the in vitro enzymatic ACE inhibitory activity data, it is evident that the synthesized compounds are not potent inhibitors of ACE enzyme. Therefore, these compounds require further modifications in their chemical structure. It is suggested that the incorporation of free amino group and free carboxylic acid groups in the structure of the synthesized compounds may provide potent and more active ACE inhibitors. Accordingly, further studies are in progress in our laboratory.
Acknowledgement
The authors would like to express their deep gratitude to the Northern Border University, Kingdom of Saudi Arabia for providing financial support (Project No. 3/3/1436/5).
References
- Gaetano S., J. Cardiovasc. Dis.2013,1(1), 1-2.
- Al-Sieni A.I., Baghdadi M.A., Al-Abbasi F.A., Asian J. Pharm. Health Sci.2014,4(1), 916-920.
- El-Bcheraoui C., Memish Z.A.,Tuffaha M.,Daoud F.,Robinson M.,Jaber S.,Mikhitarian S.,Al-Saeedi M.,Al-Mazroa M.A.,Mokdad A.H.,Al-Rabeeah A.A.,Int. J. Hypertens.2014,1-8, Article ID 564679,http://dx.doi.org/10.1155/2014/564679.
CrossRef - Aljefree N., Ahmed F., Adv. Public Health.2015, 1-23, Article ID 235101, http://dx.doi.org/10.1155/2015/235101.
CrossRef - Temilolu O.A., Miller M., World J. Cardiol.2009,1(1), 3-10.
CrossRef - Reddy K.S.,Public Health Nutr.2002,5(1A), 231-237.
- Steffen R.P., Weishaar R.E., Evans D.B., Kaplan H.R., Cardiovasc. Drug Rev.1986,4(1), 81-105.
CrossRef - Asif M., Anita S., Ovidius Univ. Ann. Chem.2011,22(2), 98-101.
- Xin-Sheng C., Hua-Wu Z., Yuang-Ying J., Wei-Qin W., Kun L., Acta Pharmacol. Sin.1990,11(4), 338-343.
- Dobariya T.D., Multani P.J., Int. J. ChemTech Res.2013,5(5), 2154-2164.
- Kauffman R.F., Utterback B.G., Robertson D.W., Circ. Res.1989,64(5), 1037-1040.
CrossRef - Nieminen M.S., Fruhwald S., Heunks L.M.A., Suominen P.K., Gordon A.C., Kivikko M., Pollesello P., Heart Lung Vessel.2013,5(4), 227-245.
- Kumar D., Carron R., De La Calle D., Jindal D.P., Bansal R., Acta Pharm.2008,58, 393-405.
CrossRef - Hiroshi M., Tohru N., Kazuhiro G., Thromb. Res.1983,31(4), 599-609.
CrossRef - Jonas R., Klockow M., Lues I., Prucher H., Schliep H.J., Wurziger H., Eur. J. Med. Chem.1993,28(2), 129-140.
CrossRef - Mochizuki N., Uchida S., Miyata H., J. Cardiovasc. Pharmacol.1993,21(6), 983-995.
CrossRef - Moore J.B.J., Combs D.W., Tobia A.J., Biochem. Pharmacol.1991,42(3), 679-683.
CrossRef - Bansal R., Thota S., Med. Chem. Res.2013,22, 2539-2552.
CrossRef - Warren S.E., Kihara Y., Pesaturo J., Gwathmey J.K., Phillips P., Morgan J.P., J. Mol. Cell. Cardiol.1989,21(10), 1037-1045.
CrossRef - Johan A.B., Richard F.W., Robert S.S., Charles K., William C.F., Henry F.C., Mark H.P., J Cardiovasc. Pharmacol.1990,16(4), 537-545.
CrossRef - Kenneth J.M., Roger J.E., John S.D., David C.G., Catherine A.S., Bella P., Aileen K., Angela W., James A.L., William J.C., Br. J. Pharmacol.1992,107, 463-470.
CrossRef - Wang T., Dong Y., Wang L., Xiang B., Chen Z., Qu L., Arzneim. Forsch.2008,58(11), 569-573.
- Somerville L.F., Allen C.F.H., Org. Syntheses, 1943,2, 81-88.
- Husain A., Sarafroz M., Ahuja P., Acta Pol. Pharm.2008,65(5), 527-534.
- Husain A., Acta Pol. Pharm.2009,66(5), 513-521.
- Brown N.J., Vaughan D.E., Circulation, 1998, 97, 1411-1420.
CrossRef - Natesh R., Schwager S.L.U., Sturrock E.D., Acharya K.R., Nature,2003,421, 551-554.
CrossRef - Loving K., Salam N.K., Sherman W.,J. Comput. Aided Mol. Des.2009,23, 541-554.
CrossRef - Lam L.H., Shimamura T., Manabe S., Ishiyama M., Ukeda H., Anal. Sci. 2008,24(8), 1057-1060.
CrossRef - Bang T.H., Suhara H., Doi K., Ishikawa H., Fukami K., Parajuli G.P., Katakura Y., Yamashita S., Watanabe K., Adhikari M.K., Manandhar H.K., Kondo R., Shimizu K., Evid. Based Complement. Alternat. Med.2014, 1-11, Article ID 195305, http://dx.doi.org/10.1155/2014/195305.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


