Antibacterial and morphological studies of electrospun silver-impregnated polyacrylonitrile nanofibre
T. O. Siyanbola1*, T. Gurunathan2, A. F. Akinsola3, J. A. Adekoya1, A. A. Akinsiku1, O. Aladesuyi1, S. Rajiv4, S. Mohanty 5, T. S. Natarajan6 and S. K. Nayak2
1Department of Chemistry, Covenant University, P. M. B. 1023, Ota, Ogun State, Nigeria.
2Central Institute of Plastics Engineering and Technology, Guindy, Chennai 600032, India.
3Department of Chemistry, Ekiti State University, P.M.B. 5363, Ado-Ekiti, Nigeria.
4Department of Chemistry, Anna University, Chennai-600025, India.
5Central Institute of Plastics Engineering and Technology, Guindy, Chennai-600032, India.
6Physics Department, Indian Institute of Technology, Madras, Chennai-600036, India.
Corresponding Authore Email: tolu.siyanbola@covenantuniversity.edu.ng
DOI : http://dx.doi.org/10.13005/ojc/320116
Article Received on :
Article Accepted on :
Article Published : 03 Mar 2016
Silver-impregnated polyacrylonitrile (PAN) nanofibre was prepared through electrospinning process. Infra-red spectroscopy (FT-IR), X-ray powder diffraction (XRD), scanning electron microscopy (SEM), Thermo gravimetric analyses (TGA/DTA) were used to characterize PAN and PAN/Ag composites. The XRD results reflects decrease in the crystallinity of PAN as it is been modified with Ag. Antibacterial activity of PAN/Ag was also investigated.
KEYWORDS:Antibacterial activity; Nanofibre, Silver; Polyacrylonitrile;Electrospinning;Composite
Download this article as:| Copy the following to cite this article: Siyanbola T. O, Gurunathan T, Akinsola A. F, Adekoya J. A, Akinsiku A. A, Aladesuyi O, Rajiv S, Mohanty S, Natarajan T. S, and Nayak S. K. Antibacterial and morphological studies of electrospun silver-impregnated polyacrylonitrile nanofibre. Orient J Chem 2016;32(1). |
| Copy the following to cite this URL: Siyanbola T. O, Gurunathan T, Akinsola A. F, Adekoya J. A, Akinsiku A. A, Aladesuyi O, Rajiv S, Mohanty S, Natarajan T. S, and Nayak S. K. Antibacterial and morphological studies of electrospun silver-impregnated polyacrylonitrile nanofibre. Orient J Chem 2016;32(1). Available from: http://www.orientjchem.org/?p=14586 |
Introduction
The undeniable contribution of electrospinning to nanotechnology development especially in the industrial and academic sectors cannot be over emphasized. It provides cheap, uncomplicated effective technology towards synthesizing nanofibre polymers with excellent properties such as flame retardation, enhanced dimensional stability, high porousity and specific surface area1-2. The high surface area property exhibited by nanofibre makes high degree of physical contact possible by creating sites for chemical reaction which is the basis for various modification of the nano fibre or trapping fine particle size material through physical entanglement thereby making filtration property of the material possible. Other areas of immense application includes tissue scaffold, material for dressing wounds, sensors, storage cells for hydrogen fuel cells, drug delivery systems, catalysis reaction and protective clothing etc3-5. Typical electrospinning procedure involves the use of high voltage source that provides the required electric field on the polymer solution or melts which in turn jets out of the syringe. The dry jets nanofibre move towards an earmark opposite charge collector which capture the polymer fibre (Figure 1). The process is void of coagulation chemistries or high temperatures that can produce solid threads from solutions. The type of polymer solution and/or spinneret modification will determine the type of nanofibre that is will be formed. Sometimes, the nanofibres can exhibit porous or core-shell morphology depending on the material undergoing spinning, nozzle to collector distance as well as its miscibility and rate of evaporation of the solvent involved6. Megelski et al., (2002) reported bead formation morphology in electrospun polystyrene fibre but on decreasing nozzle to collector plate distance but on increase the nozzle to collector plate distance, the ribbon shaped morphology is conserved7. It is however important to state that alteration of the distance between the feeding nozzle and the fibre collecting device gives more drying time to the electrospun fibres. Several base polymeric materials such as collagen, polylactide-glycolide, poly(ethylene oxide), silk protein, cellulose derivatives and polyvinyl alcohol (PVA) have been used as precursors8.
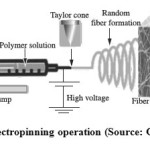 |
Figure 1a: Electropinning operation (Source: Google image9) |
The impregnation of inorganic nanoparticles into polymer matrix has been synthetically achieved recently10. Nanoparticles such as metal oxide, metal, metal sulphide, metal nitrate and so on. The internalization of nano-AgX into polymeric matrix offers the composite extensive industrial viabilities that combine features of organic and inorganic materials11-12. They provide composite materials having antimicrobial, optical and electrical applications10, catalyst and photochromic materials13-14.
Experimental
Polyacrylonitrile (PAN) and silver nitrate was purchased from Sigma Aldrich, India. DMF was obtained from Sisco Research Laboratory Private Limited, India.
Solution preparation
PAN solution and PAN/AgNO3 solution were both separately prepared by dissolving them in DMF and respectively stirred continuously for 8 and 24 hours on a magnetic stirrer.
Preparation of dispersed Ag nanoparticle in PAN nano-fibrous film
A 10×20 mm nano-fibrous film of PAN/ AgNO3 was place in a 50 mL beaker and 20 mL triple distilled water plus 1 mL N2H5OH aqueous solution was added to it. This reduces the silver ions in PAN nanofibrous film to silver at room temperature. After 30 minutes, the film was washed with distilled water and alcohol. After which, it was dried in a vacuum oven.
Electrospinning of PAN/Ag
Figure 1 shows the set-up used for electrospinnig of PAN/Ag. The polymer solution of PAN/Ag was taken into a 2 mL syringe connected to a capillary tip of 0.56 mm inner diameter. The positive electrode to which high voltage power supply is connected is wrapped with aluminium foil. Optimization of electric voltage was at 15 KV. Different polymer solutions concentrations were electrospun at 0.7 µL/min flow rate and 10 cm tip to collector distance. All experiments were carried out at 25 ºC.
Characterization
Fourier Transform Infrared Spectroscopy (FT-IR) analysis
The functional groups present in the spun fibres were identified by Bruker 66V FT-IR spectrometer in the range 4000-400 cm-1.
Scanning electron microscopy (SEM) analysis
Eletrospun fibre mats were sputter coated with gold and the micro to nano-fibres morphology were observed on a high resolution scanning electron microscope (HRSEM). Scion image software was used analyzing the fibre diameters, orientation pattern and surface of the individual fibre.
X-Ray diffraction (XRD)
The X-ray diffraction measurements of nanofibre were performed on Rigaku Miniflex II instrument. The nanofibrous membrane was pressed inside the diffractometer sample holder and patterned data were collected in-situ.
Thermal analysis
The thermal stability of PAN and PAN/Ag nano-fibrous materials were ascertained by TGA/DTA analysis. Samples weight ranging from 4-8 mg was placed in the platinum sample pans under a continuous nitrogen atmosphere with a flow of 25 mL/min in a TGA/DTA model SDT 2600 instrument. The sample and its reference were heated at the rate 20 ºC/min. The phase changes and glass transition temperature of the nanofibrous Pan and PAN/Ag were measured on Perkin-Elmer differential scanning calorimeter (DSC-2).
Antibacterial activity
The standard LB plate method was used to evaluate the antibacterial activity of the AgNO3/PAN precursor solution against gram negative Escherichia coli (E. coli). The ready-made nutrient agar was suspended in double distilled water (1000 mL) and heated to boiling for 25 min until it dissolved completely; the medium and Petri dishes were autoclaved at pressure 15 Lb/in2. The medium was transferred into sterile Petri dishes under aseptic conditions in a laminar air flow chamber. When the medium in the plates had solidified, 0.5 mL (approx. 106 CFU/mL) of culture of test organism was inoculated and evenly spread over the agar surface with a sterile L-shaped rod. Embedded nanofibrous samples with 2 cm × 2 cm (approx.) samples were cleaned with double distilled water and placed on the medium and incubated at 37 °C for 24 hours. The antibacterial activity was carried out based on the formation of an inhibition zone and loss of growth of organisms beneath and surrounding the films placed on the agar medium. Three replicates were used for each treatment15.
Results and Discussion
Fourier Transform Infra-red Spectroscopy
It had been reported that electropinning process of composite solutions might lead to differences in the composition of the pre-spun solution and the nanofibres deposited12-13. In the light of this, the infra-red spectroscopy of deposited fibres was analyzed. The overlay FT-IR spectra of PAN and PAN/Ag are represented in Figure 1. PAN shows a characteristic stretching vibrational peak of nitrile group (-CN-) at 2244 cm-1. Also, stretching and bending vibrations of methylene (-CH2-) functional group is seen at 2917 cm-1 and 1448 cm-1 respectively. As for PAN/Ag nanofibre, the peak at 1637 cm-1 is due to the oxidation of the PAN in air (as obtained), which results in the formation of carboxyl (C=O) groups. The peaks at 1248 cm-1 and 1352 cm-1 are assigned to the aliphatic -CH- group vibrations of different modes in CH and CH2, respectively. The positions of these peaks in PAN/Ag composites nanofibre is observed to shift slightly lower values due to the interaction between PAN molecules and Ag.
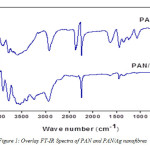 |
Figure 1b: Overlay FT-IR Spectra of PAN and PAN/Ag nanofibres |
X-ray diffraction analysis (XRD)
The representative XRD patterns of PAN and PAN/Ag fibres are presented in figure 2. The PAN single unique diffraction peak is observed at 2θ = 17° while the modified electrospun nanofibre (PAN/Ag) shows other characteristic peaks at 38°, 44°, 64°, and 77° of 2θ.
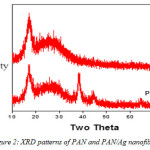 |
Figure 2: XRD patterns of PAN and PAN/Ag nanofibres |
Scanning electron microscopy (SEM)
Figure 3 shows the SEM image of PAN electrospun nanofibre at 12 wt% concentration and at 5 µm (3a) and 10 µm (3b) magnifications. The mean diameters of the fibres were found to be approximately 150 nm to 1nm respectively. Figure 4 shows the histogram peaks for electrospun PAN nanofibres.
 |
Figure 3: SEM pictures of electrospun PAN nanofibre |
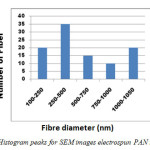 |
Figure 4: Histogram peaks for SEM images electrospun PAN nanofibers |
Thermogravimetric Analysis (TGA) and Differential Thermal Analysis (DTA)
The TGA and DTA thermograms of PAN and PAN/Ag fibres are presented in Figure 5. Both thermograms show evident weight loss peaks at 305-320 ºC and 600-740 ºC in the TGA curves. The first weight loss is presumed to be caused by decarboxylation of the methylacrylate in PAN while, the second weight loss may be due to thermo-oxidative degradation of PAN macromolecular chains. For PAN/Ag nanocomposite, the first step degradation was observed at around 325 ºC whereas, that of PAN was at 309 ºC. The PAN/Ag system is seen to be more thermally stable that PAN system.
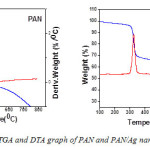 |
Figure 5: TGA and DTA graph of PAN and PAN/Ag nanofibres
|
Antibacterial activity
The antibacterial activity of PAN/Ag nanocomposite fibre is tested against Gram-negative Escherichia coli. The test was carried out with varying percentage of Ag+ in PAN solution. As the percentage composition increases the zone of inhibition also increases (Figure 6). The formation of clear zone of inhibition around the PAN/Ag sample clearly shows that the Ag+ ions were effective on the inhibition of bacterial growth.
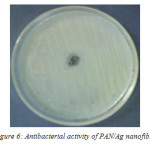 |
Figure 6: Antibacterial activity of PAN/Ag nanofibre |
Conclusion
Electrospun PAN and PAN/Ag nanofibres were characterized by FT-IR, XRD, SEM, TGA and DTA. Antibacterial activity of PAN/Ag was also evaluated. The SEM micrographs show 100 % bead free nanofibre in 12 wt% ratio of dimethylformaldehyde (DMF). The FT-IR absorption peaks and XRD pattern of PAN/Ag clearly reveal the interaction of Ag with PAN molecules
Reference
- Pham, Q. P.; Sharma, U.; Mikos, A. G. Tissue Engineering. 2006, 12, 1197-1211.
CrossRef - Park, J. H.; Karim, M. R.; Kim, I. K.; Cheong, I. W.; Kim, J. W.; Bae, D. G.; Cho, J. W.; Yeum, J. H. Colloid Polm. Sci. 2010, 288, 115-121.
CrossRef - Reneker, D. H.; Chun, I. Nanotechnology. 1996, 7, 216-223.
CrossRef - Li, D.; Xia, Y. Adv. Mater. 2004, 16, 1151-1170.
CrossRef - Son, W. K.; Youk, J. H.; Lee, T. S.; Park, W. H. Polym. 2004, 45, 2959-2966.
CrossRef - Bazilevsky, A. V.; Yarin, A. L.; Megaridis, C. M. Langmuir. 2007, 23, 2311-2314.
CrossRef - Megelski, S.; Stephens, J. S.; Chase, D. B.; Rabolt, J. F. Macromol. 2002, 22, 8456-8466.
CrossRef - Won, H. P.; Lim, J.; Dong, I. Y.; Hudson, S. Polym. 2004, 45, 7151-7157.
CrossRef - Dhandayuthapani, B.; Yoshida, Y.; Maekawa, T.; Kumar, D. S. Mat. Res. 2011, 14, 317-325.
CrossRef - Chan, Y.; Liu, H.; Zhang, Z. Appl. Mechan. & Mater. 2010, 26-28, 159-162.
CrossRef - Mbhele, Z. H.; Salemane, M. G.; Van Sittert, C. Chem. Mater. 2003, 26, 5019-5024.
CrossRef - Kim, J. H.; Min, B. R.; Won, J.; Joo, S. H.; Kim, H. S.; Kang, Y. S. Macromol. 2003, 36, 6183-6188.
CrossRef - Currao, A.; Reddy, V .R.; Calzaferri, G. Chem. Phys. Chem. 2004, 5, 720-724.
CrossRef - Schurch, D.; Currao, A.; Sarkar, S.; Hodes, G.; Calzaferri, G. J. Phys. Chem. B. 2002, 106, 12764-12775.
CrossRef - Siyanbola, T. O.; Sasidhar, K.; Rao, B. V. S. K.; Narayan, R.; Olaofe, O.; Akintayo, E. T.; Raju, K. V. S. N. J. Am. Oil Chem. Soc. 2015, 92, 267-275.
CrossRef - Wongsasulak, S.; Kit, K. M.; McClements, D. J.; Yoovidhya, T.; Weiss, J. Polym. 2007 48, 448-457.
CrossRef - Kriegel, C.; Kit, K. M.; McClements, D. J.; Weiss, J. Polym. 2009, 50, 189-200.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


