DFT investigation on the tautomerization reaction and NBO analysis of the Acamprosate drug
B. Esmaeili1, S. A. Beyramabadi*,1,2, R. Sanavi-Khoshnood1, A. Morsali1,2
1Department of Chemistry, Mashhad Branch, Islamic Azad University, Mashhad, Iran 2Research Center for Animal Development Applied Biology, Mashhad Branch, Islamic Azad University, Mashhad 917568, Iran Corresponding Author Email :: beyramabadi@yahoo.com
DOI : http://dx.doi.org/10.13005/ojc/310434
Article Received on :
Article Accepted on :
Article Published : 10 Nov 2015
The Acamprosate is a significant drug for alcohol abuse therapy, which may be an effective treatment for tinnitus, too. The Acamprosate has two possible tautomers, Keto and Enol tautomers. Each of the tautomers involves two important conformers. In this work, employing density functional theory (DFT) and handling the solvent effects with the PCM model, the structural parameters, energetic behavior, natural bond orbital analysis (NBO), as well as tautomerism mechanization of the Acamprosate are investigated in several solvents. The conformers of the Keto tautomer are more stable than the corresponding conformers of the Enol tautomer. A strong intramolecular hydrogen bond stabilizes the molecule significantly. Increasing the polarity of the solvent reduces the rate of the tautomerization of the Acamprosate by increasing the Ea of the reaction. A large HOMO-LUMO energy gap for the most stable tautomer implies high stability of the Acamprosate.
KEYWORDS:Acamprosate;DFT; PCM; Tautomerism; Hydrogen Bond; NBO; Tautomer
Download this article as:| Copy the following to cite this article: Esmaeili B, Beyramabadi S. A, Sanavi-Khoshnood R, Morsali A. DFT investigation on the tautomerization reaction and NBO analysis of the Acamprosate drug. Orient J Chem 2015;31(4). |
| Copy the following to cite this URL: Esmaeili B, Beyramabadi S. A, Sanavi-Khoshnood R, Morsali A. DFT investigation on the tautomerization reaction and NBO analysis of the Acamprosate drug. Orient J Chem 2015;31(4). Available from: http://www.orientjchem.org/?p=12538 |
Introduction
The Acamprosate or calcium –N–acetylhomtaurinate is a drug successfully used for alcohol abuse therapy in maintaining abstinence following alcohol withdrawal [1-8]. The Acamprosate has been available in several European countries for many years that was approved by the US Food and Drug Administration (FDA) in 2004 for use in alcoholic individuals to decrease alcohol craving after alcohol detoxification [9]. Also, some studies show that the Acamprosate is an effective treatment for tinnitus, persistent ringing in the ears due to hearing loss or benzodiazepine withdrawal [10]. However, mechanism of action of the Acamprosate is unknown and arguable [1-2,11].
Now, the DFT methods provide valuable information in many areas of the computational chemistry. For example, structural parameters, spectroscopic assignments, kinetics and mechanism investigations of the chemical reactions, drug science and so on [e.g. 12-26].
The knowledge of the structural parameters of drugs is an essential prerequisite for better understanding of their biological activities. So far, no crystal structure has been reported for the Acamprosate and no theoretical study has been published on this drug. Since, a detailed computational investigation on the Acamprosate is of major importance. Herein, we address this issue and examine its geometry, stability, tautomerization reaction and the natural bond orbital (NBO) analysis using the DFT approaches.
Theoretical Methods
In this work, all of the calculations have been have been performed by using the hybrid functional (B3LYP)[27] as implemented in the Gaussian 03 software package [28]. The 6-311+G (dip) basis set was employed.
Solvent plays important role in chemical reactions. Here, the sophisticated Polarizable Continuum Model (PCM) [29] has been employed for investigation of solute-solvent interactions in water, methanol, ethanol and acetone solutions.
In each of the solvents, all degrees of freedom for all geometrics were fully optimized. The optimized geometries were confirmed to have no imaginary frequency, except for transition state (TS) that has only one imaginary frequency of the Hessian. The zero-point corrections and thermal corrections have been considered in evaluation of the energies.
Results and Discussion
Molecular Geometry
The Acamprosatedrug could exist as two possible tautomers; Keto and Enol tautomers. Each of the Keto and Enol tautomers has two conformers; geometries of which have been fully optimized in four different solvents using the PCM model. The PCM optimized geometries of them are shown in Fig. 1. Two conformers of the Keto tautomer are named as A1 and A2, where two conformers of the Enol tautomer are named as A3 and A4, respectively.
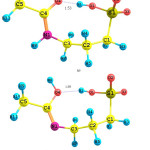 |
Figure 1: Optimized geometries for the A1-A4 species of the Acamprosate. |
The relative energies of the optimized geometries are gathered in Table 1, where the zero-point corrections have been considered. As seen the A2 is the most stable structure of the Acamprosate in all of the investigated solutions.
Table 1: The PCM relative energies (KJ.mol-1) of the A1 to A4 species
|
Species |
Acetone |
Ethanol |
Methanol |
Water |
|
|
A2 |
0.0 |
0.0 |
0.0 |
0.0 |
|
|
A3 |
19.44 |
19.28 |
19.08 |
26.65 |
|
|
A1 |
97.97 |
98.28 |
98.60 |
99.01 |
|
|
A4 |
97.12 |
97.18 |
97.64 |
97.92 |
|
|
TS2-3 |
207.21 |
207.38 |
207.54 |
211.30 |
|
|
|
|||||
Two conformers of the Keto tautomer are A1 and A2. There is an intramolecular-hydrogen bond in the optimized geometry of theA2 conformer, which increases stability of the A2 conformer by 99.01 kJ.mol-1 in comparison with the A1 conformer, in the aqueous solution. The H-bond forms between the H1 atom and O4 atom of the carbonyl group. The O1H1…O4 distance and O1-H1-4 angle are 1.53 Ǻ and 175.5°, respectively.
For the Enol tautomer, there is an intramolecular-hydrogen bond in the A3 conformer, too, stabilizes the A3 conformer than the A4 one by 5.09 kJ.mol-1 in the aqueous solution. In the optimized geometry of the A3 conformer, the O1H1…O4 distance and O1-H1-4 angle of the H-bond are 1.71 Ǻ and 107.4°, respectively.
Table 2: Selected structural parameters for the optimized geometries in aqueous solution.
|
A1 form |
A2 form |
A3 form |
A4 form |
TS 2-3 |
|
|
Bond length (Ǻ) |
|||||
|
N1-H8 |
1.00 |
1.00 |
3.10 |
3.09 |
1.34 |
|
N1-C3 |
1.45 |
1.46 |
1.46 |
1.46 |
1.46 |
|
N1-C4 |
1.35 |
1.34 |
1.26 |
1.26 |
1.32 |
|
C4-O4 |
1.23 |
1.24 |
1.38 |
1.37 |
1.29 |
|
C5-H9 |
1.09 |
1.09 |
1.09 |
1.09 |
1.09 |
|
C3-C2 |
1.53 |
1.54 |
1.54 |
1.53 |
1.54 |
|
C2-C1 |
1.52 |
1.53 |
1.53 |
1.52 |
1.53 |
|
C1-S1 |
1.81 |
1.81 |
1.81 |
1.81 |
1.81 |
|
S1-O1 |
1.63 |
1.59 |
1.62 |
1.63 |
1.61 |
|
S1-O2 |
1.45 |
1.46 |
1.46 |
1.45 |
1.46 |
|
O1-H1 |
0.97 |
1.02 |
0.98 |
0.97 |
0.99 |
|
O4-H8 |
3.15 |
3.15 |
0.96 |
0.96 |
1.31 |
|
Angle (˚) |
|||||
|
N1-C4-H8 |
118.3 |
118.2 |
148.9 |
149.7 |
53.9 |
|
N1-C4-O4 |
122.5 |
122.4 |
121.4 |
121.8 |
106.5 |
|
C4-C5-H9 |
108.7 |
108.7 |
110.5 |
110.4 |
108.5 |
|
C4-C5-H10 |
108.7 |
108.7 |
110.5 |
110.5 |
109.9 |
|
N1-C3-H6 |
107.7 |
107.9 |
111.09 |
112.6 |
106.6 |
|
N1-C3-H7 |
108.1 |
107.9 |
107.7 |
107.3 |
109.1 |
|
C3-H6-H7 |
107.9 |
106.8 |
106.8 |
107.6 |
106.8 |
|
C2-H4-H5 |
106.7 |
105.8 |
105.8 |
106.6 |
105.8 |
|
C1-S1-O1 |
104.2 |
104.6 |
104.6 |
104.4 |
103.7 |
|
C1-S1-O2 |
110.1 |
108.6 |
109.1 |
110.2 |
109.5 |
|
C1-S1-o3 |
108.7 |
108.8 |
109.5 |
108.7 |
109.5 |
|
Dihedral angle (˚) |
|||||
|
C5-C4-N1-O4 |
179.7 |
-179.9 |
-179.9 |
-179.7 |
-179.6 |
|
C4-C5-H9-O4 |
-130.1 |
-130.3 |
26.6 |
-125.4 |
26.1 |
|
H8-N1-C2-O4 |
140.7 |
178.8 |
178.6 |
146.3 |
106.7 |
|
N1-H8-C4-C3 |
176.8 |
-178.7 |
0.96 |
1.8 |
-127.3 |
|
C1-S1-O3-O2 |
127.4 |
-124.8 |
-126.6 |
-128.0 |
-126.8 |
Tautomerization Mechanism
The most stable conformer for the Keto and Enol tautomers of the Acamprosate drug are the A2 and A3 species, respectively. In both of the A3 and A2 species, there is an intramolecular-hydrogen bonding between the H1 and O4 atoms, increasing stability of them. Herein, the Enol-Keto tautomerization of the Acamprosate has been investigated as the A2-A3 tautomerization in details. The solvent effects have been considered in water, methanol, ethanol and acetone solutions.
Going from the A2 tautomer to the A3 one, the H8 atom is transferred from the N1 atom to O4 atom. The obtained structure for the transition state of this reaction (TS2-3)is shown in Fig.4. Important structural parameters of the TS2-3 are gathered in Table 2 together with the A1 to A3 tautomers for comparison.
In the optimized geometry of the TS2-3, breaking of the N1-H8 bond together with the formation of O4-H8 bond is clear. The N1-H8 and O4-H8 distances vary from 1.00 and 3.15 Ǻ for the A2 tautomer to 1.34 and 1.31 Ǻ for the TS2-3, respectively. These distances are 3.10 and 0.96 Ǻfor the A3 tautomer, respectively.
As seen in Table 1, going from the non-polar solvent to the polar solvent (in the water, methanol, ethanol and acetone order)barrier energy (Ea) of the A2-A3 tautomerization is increased slightly. The largest Ea is related to the aqueous solution. Increasing of the polarity of the solvent stabilizes the A2 species more than the other investigated conformers of the Acamprosate, too.
Considering the equilibrium between the A2 and A3 tautomers, the value of the tautomeric equilibrium constant (K) is calculated by using

where ∆G, R and T are the Gibbs free energy difference between the two tautomers, the gas constant and temperature, respectively.
In the aqueous solution phase, the ∆G between the A2 and A3 tautomers is 99.14 kJ.mol-1, in favor of the A2 tautomer. Hence, using the Eq(1), amount of the A3 tautomer in aqueous solution of the Acamprosate is predicted to be negligible; only 0.4%. Also, the ∆G between the A2 and A1 conformers of Keto tautomer is 18.57 kJ.mol-1, in favor of the A2 tautomer. Therefore the amount of the A1 conformer in aqueous solution is predicted to be negligible, too.
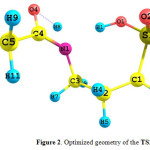 |
Figure 2: Optimized geometry of the TS2-3. |
Nbo Analysis
The NBO analysis is used as an useful tool for investigation of the intra- and intermolecular bonding interactions and investigation of charge transfer in chemical compounds [20,30,31]. Electron delocalization between donor NBO(i) and acceptor NBO(j) orbitals causes to the stabilization energy of hyper conjugative interactions (E(2)). Amount of the E(2) is a criteria for determining degree of interaction between electron donor and electron acceptor orbitals. The greater the E(2), the greater electron transferring tendency from electron donor to electron acceptor, resulting in more electron density delocalization, and consequently leading to more stabilization of the system. The value of E(2) is calculated by using [20,31,32]

where qi, Fij, εj and εi parameters are the donor orbital occupancy, the off-diagonal NBO Fock matrix element, energies of the acceptor and donor orbitals, respectively. The lower εj-εi energy difference causes to higher the E(2) stabilization energy. The parameters of Eq. (2) have been obtained from the second-order perturbation theory analysis of Fock matrix in NBO basis. The selected results for the most stable tautomer of the Acamprosate, A2, are listed in Table 4, where the Fij is Fock matrix element between i and j NBO orbitals.
Table 4: The second-order perturbation theory analysis of Fock matrix in NBO basis for the A2tautomer of the Acamprosate.
| Donor NBO (i) | Acceptor NBO (j) |
Ej-Ei(a.u.) |
Fij(a.u.) |
E(2) (kcal/mol) |
| BD(2) N1-H8 | BD*(1) C4-O4 | 1.01 | 0.059 |
4.01 |
| LP(2) O1 | BD*(1) S1-O2 |
0.62 |
0.055 |
5.95 |
| LP(2) O1 | BD*(1) S1-C1 |
0.47 |
0.049 |
6.05 |
| LP(2) O2 | BD*(1) S1-O3 |
0.58 |
0.063 |
8.44 |
| LP(2) O2 | BD*(1) S1-C1 |
0.43 |
0.074 |
15.90 |
| LP(3) O2 | BD*(1) S1-O1 |
0.42 |
0.091 |
24.00 |
| LP(3) O2 | BD*(1) S1-O3 |
0.58 |
0.074 |
11.47 |
| LP(2) O3 | BD*(1) S1-O2 |
0.58 |
0.077 |
12.83 |
| LP(2) O3 | BD*(1) S1-C1 |
0.43 |
0.070 |
14.43 |
| LP(2) O3 | BD*(1) S1-O1 |
0.42 |
0.092 |
24.85 |
| LP(2) N1 | BD*(2) C4-O4 |
0.45 |
0.101 |
27.26 |
| LP(2) O4 | BD*(1) C4-N1 |
1.11 |
0.081 |
7.43 |
| LP(2) O4 | BD*(1) O1-H1 |
0.73 |
0.154 |
39.77 |
| LP(2) O4 | BD*(1) C4-C5 |
0.77 |
0.097 |
14.57 |
Based on the DFT results, the strongest interaction inducing the highest stabilization energy is the n(O4) →σ*(O1-H1) electron donations. Since, strong O1H1…O4 intramolecular hydrogen bond has significant role in stabilization of the Acamprosate by 39.77 kcal/mol.
There are several electron donations from lone pair electrons of oxygen atoms of the –SO3H moiety to the antibonding orbitals of the S-O and S-C bonds, which stabilize the molecule (Table 4).
The spatial distribution for the highest-occupied-molecular orbital (HOMO) and the lowest-unoccupied-molecular orbital (LUMO) of the A2 tautomer are shown in Fig. 3. The HOMO orbital is localized on theO4 and N1 atoms. But the LUMO orbital is mainly localized on C4-O4 and C4-N1 bonds. As seen in Table 4, electron transitions from the O4 and N1 atoms to the C4-O4 and C4-N1 bonds (HOMO to LUMO electron transitions) stabilize the Acamprosate significantly.
The HOMO-LUMO energy gap is one of the important characteristics of molecules, which has a majorrole in such cases as electric properties, electronic spectra and photochemical reactions. The value of the energy gap between the HOMO and LUMOfrontier orbitals of the A2tautomer ofthe Acamprosateis equal to 6.86 eV. This large energy gap confirms that structure of the Acamprosate drug is very stable [32,33].
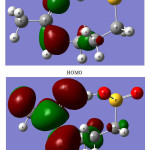 |
Figure 3: The HOMO and LUMO frontier orbitals of the A2 tautomer of Acamprosate. |
Conclusion
The Acamprosate could exist as two possible tautomers, Keto and Enol form. For each of the tautomer, two conformers have been optimized, A1 and A2 for the Keto tautomer, A3 and A4for the Enol one.Herein, optimized geometries and energetic characters of the possible conformers of the tautomers, as well as kinetic and mechanism of the Acamprosate tautomerization have been investigated in detail using DFT approaches.
Because of the intramolecular hydrogen bond,theA2 and A3conformers of the Keto and Enol tautomersare more stable than the A1 and A4 conformers, respectively.In overall, the Keto form is more stable than the Enol form. The A2 is the most stable tautomer of Acamprosate. This tautomer can be converted to the A3 tautomer via an IPT. The A2⇄A3 tautomerism has been investigated in several solvents. Higher polarity of the solvent stabilizes the A2 form more than the other optimized geometriesof Acamprosate, increases the Ea of the A2⇄ A3 tautomerization, too. The Ea of the reaction is 211.30 kJ/mol in aqueous solution.
The NBO results shown that thestrong intramolecular hydrogen bond in the A2 formstabilizes the molecule significantly by 39.77 kcal/mol.The HOMO and LUMO frontier orbitals of the A2are localized mainly on the O4-C4-N1 region. The large HOMO-LUMO energy gap indicates high stability of the A2 tautomer.
References
- Courtyn, J., Cornelissen, B., Oltenfreiter, R., Vandecapelle, M., Slegers, G. and Strijckmans, K.Nucl. Med. Biol. 2004, 31, 649-654.
- Courtyn, J., Goethals. P., Eycken. J.V. and Dams, R.J. Labelled. Comp. Radiopharm.2001, 44, 643-651.
- Carmen, B., Angeles, M., Ana, M. and María. A.J. Addiction2004, 99, 811-828.
- Kranzler, H. R. and Kirk, J. Alcohol. Clin. Exp. Res.2001, 25, 1335-1341.
- Mann, K., Lehert, P. and Morgan, M.Y. 2004, 28, 51-63.
- Sass, H., Soyka, M., Mann, K. and Zieglgänsberger, W. Arch. Gen. Psychiat.1996, 53, 673-680.
- Zeise, M.L., Kasparov, S., Capogna, M. and Zieglgänsberger, W. Eur. J. Pharm.1993, 231, 47-52.
- Mann, K., Kiefer, F., Spanagel, R. and Littleton J. Alcohol. Clin. Exp. Res.2008, 32, 1105-. 1110.
- Booth by, L. A. and Doering, P. L. Clin. Ther.2005, 27, 695-714.
- Azevedo, A. A. and Figueiredo, R. R. Rev. Bras. Otorrinolaringol.2005, 71, 618-623.
- Reilly, M.Y., Lobo. I.A., McCracken, L.M., Borghese, C.M., Gong, D., Horishita, T., and Harris, R.A. Alcohol. Clin. Exp. Res.2008, 32, 188-196.
- Beyramabadi, S.A., Eshtiagh-Hosseini, H., Housaindokht, M.R. and Morsali, A. Organometallics2008, 27, 72-78.
- Galasso, V. Chem. Phys. Lett.2009, 472, 237-242.
- Eshtiagh-Hosseini, H., Beyramabadi,S.A., Mirzaei,M., Morsali,A., Salimi,A.R. and Naseri, AJ. Struct. Chem.2013, 54, 1063-1069.
- Beyramabadi,S.A., Morsali,A. and Shams, A. J. Struct. Chem.2015, 56, 243-249.
- Eshtiagh-Hosseini, H., Beyramabad, S..A., Morsali, A., Mirzaei, M., Chegini, H., Elahi, M. and Naseri, M.A. J. Mol. Struct.2014, 1072, 187-194.
- Eshtiagh-Hosseini, H., Housaindokht, M.R., Beyramabadi, S.A., Mir Tabatabaei, S.H., Esmaeili, A.A. and Javan-Khoshkholgh, M. Spectrochim. Acta Part A2011, 78, 1046-1050.
- Beyramabadi, S.A., Morsali, A., Javan-Khoshkholgh, M. and Esmaeili, A.A. J. Struct. Chem.2012, 53, 460-467.
- Beyramabadi, S.A., Morsali, A., Javan-Khoshkholgh, M. and Esmaeili, A.A. Spectrochim. Acta Part A2011, 83, 467-471.
- Sadeghzade, Z., Beyramabadi, S.A. and Morsali, A. Spectrochim. Acta Part A2015, 138, 637-642.
- Naghsh, F. Orient. J. Chem.2015,31, 465-478.
- Darzi, N.; Morsali, A.; Beyramabadi, S. A.Orient. J. Chem.2015,31, 1121-1125.
- Yousefian, Z. Orient. J. Chem. 2014,30, 1681-1693.
- Beyramabad, S.A., Morsali A. and Elahi, M. Prog. React. Kin. Mech.2015, Accepted for publication.
- Rahbar M., Morsali A., BozorgmehrM.R., BeyramabadiS.A.Orient. J. Chem.2015,31, Accepted for publication.
- MansoorinasabA., MorsaliA., HeraviM.M., BeyramabadiS.A. Orient. J. Chem.2015,31, Accepted for publication..
- Lee, C., Yang, W. and Parr, R.G. Phys. Rev. B1988, 37, 785-789.
- M.J. Frisch, et al. Gaussian 03, Revision B.03; Gaussian, Inc.: Pittsburgh PA, 2003.
- Cammi, R. and Tomasi, J. J. Comput. Chem.1995, 16, 1449-1458.
- Snehalatha, M., Ravikumar, C., Hubert Joe, I., Sekar, N.and Jayakumar, V.S. Spectrochim. Acta Part A2009, 72, 654-662.
- Tezer, N. and Karakus, N. J. Mol. Model.2009, 15, 223-232.
- Schwenke, D.W. and Truhlar, D.G. J. Chem. Phys.1985, 82, 2418-2427.
- Özdemir, N., Dinçer, M., Çukurovah, A. and Büyükgüngör, O. J. Mol. Model.2009, 15, 1435-1445.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


