Synthesis and Anti-Cancer Studies of 2, 6-Disubstituted Benzothiazole Derivatives
Gnanavel Sadhasivam1*, Kannan Kulanthai1, Adhirajan Natarajan2
1Department of Chemistry, Government College of Engineering, Salem-636011, Tamil Nadu, India. 2Pharmaceutical Biotechnology, KMCH College of Pharmacy, Kalapatty Road, Coimbatore-48, Tamil Nadu, India. E-mail:gnanamphd@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/310224
Article Received on :
Article Accepted on :
Article Published : 01 Jun 2015
Missing
KEYWORDS:Benzothiazole; Anticancer; MCF-7; HeLa; MG63
Download this article as:| Copy the following to cite this article: Sadhasivam G, Kulanthai K, Natarajan A. Synthesis and Anti-Cancer Studies of 2, 6-Disubstituted Benzothiazole Derivatives. Orient J Chem 2015;31(2). |
| Copy the following to cite this URL: Sadhasivam G, Kulanthai K, Natarajan A. Synthesis and Anti-Cancer Studies of 2, 6-Disubstituted Benzothiazole Derivatives. Available from: http://www.orientjchem.org/?p=8925 |
Introduction
The development of new anticancer therapeutic agents is one of the fundamental goals in medicinal chemistry. A dose of anticancer drug sufficient to kill tumour cells is often toxic to the normal tissue and leads to many side effects, which in turn, limits its treatment efficacy. In recent years, there has been a concerned search for the discovery and development of novel selective anti-tumor agents.
Benzothiazoles are bicyclic ring system with multiple applications. 2-substituted benzothiazole scaffold is one of the privileged structure in medicinal chemistry (1, 2) and reported cytotoxic on cancer cells(3-5). In a recent years number of such derivatives have exhibited interesting anticancer activities(6-11). Among the anti-tumor drugs discovered in the recent years, various benzothiazoles (12-14) possess potent anticancer properties.
On the basis of exhaustive literature review, it has been found that benzothiazole derivatives have good potential to exhibit anticancer activity (15-17). So the present study involves the syntheses of 2,6-disubstituted benzothiazole followed by preliminary cytotoxicity screening against three human cancer cell lines( MCF-7, HeLa and MG63) using MTT assay at 48 h of exposure.
Material and Methods
Chemistry
The synthetic starting material, reagents, and solvents were of analytical reagent grade or the highest quality commercially available and were purchased from sigma-Aldrich Chemical Co., Merck Chemical Co. Melting points were recorded by labtronics digital melting point apparatus. The 1H-NMR and 13C-NMR spectra were recorded in DMSOd6 solvent on Bruker 300 MHz spectrophotometer using tetramethylsilane as an internal reference, respectively. The apparent resonance multiplicity is described as s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet) and m(multiplet). Infrared measurements were recorded in the range 400–4000 cm_1 by Perkin Elmer. Elemental analysis was carried out using Perkin Elmer CHNS. Mass spectra were recorded on a Thermo LCQ Deca XP MAX at 70eV. Thin layer chromatography (TLC) analysis was carried out on 5×20 cm plate coated with silica gel GF254.
Synthesis of N-(6-nitro-1,3-benzothiazol-2-yl)acetamide (1)
To a solution of 2-amino-6-nitrobenzothiazole(10 g,0.051 mol) in 40ml acetic anhydride, pyridine(12.4 ml,0.153 mol) was added. The reaction mixture was heated to 90o C for 4 hr,and then allowed to cool. The reaction was monitored by TLC. The reaction mixture was poured on to 200 ml of 2N HCl. The solid product that formed was collected by filtration and washed with water and diethyl ether finally dried to give compound 1. Pale yellow solid ; Yield 79%; m.p. 285-286oC;IR (KBr) νmax in cm-1: 3387 (NH), 3091, 2944 (CH ), 1701(C=O), 1554 (NH bend), 1514, 1340 (NO2), 1267 (C-N ), 750 (CH bend); 1H NMR (DMSO-d6) δ ppm: 2.23 (s, 3H), 7.84 (d, 1H, J= 9 Hz), 8.23 (dd, 1H, J=2.4, 9 Hz), 8.98 (d, 1H, J=2.1 Hz), 12.72 (s, 1H, NH); 13C NMR (DMSO-d6) δ ppm: 22.7, 118.8, 120.4, 121.6, 132.0, 142.8, 153.3, 163.3, 170.1; LC-MS (ESI) m/z : 236.20 (M-H)–.
Synthesis of N-(6-amino-1,3-benzothiazol-2-yl)acetamide (2)
A solution of compound 1(10g,0.0421 mol) in 80 ml 12N HCl ,SnCl2 .2H2O(47.4g,0.210 mol) was added at RT. The reaction mixture was stirred at RT for 2 hr. The reaction was monitored by TLC. The reaction mixture was diluted with 300 ml of cold water. The solution was basified to pH 9 with 40% NaOH and the aqueous layer was extracted with ethyl acetate (3×150 mL),washed with water (2×20 mL), Brine(1x250mL) and dried over anhydrous Na2SO4. the solvent was concentrated under reduced pressure to give the compound 2. Off-white solid; Yield 82%; m.p 248-249 oC; IR (KBr) νmax in cm-1: 3417, 3298 (NH), 3057, 2916 (CH), 1692 (C=O), 1608 (C=N), 1556 (NH bend), 1272 (C-N),694 (CH); 1H NMR (DMSO-d6) δ ppm: 2.14 (s, 3H), 5.13 (s, 2H), 6.69 (dd, 1H, J =2.1, 8.4 Hz),6.99 (d, 1H, J =2.1 Hz), 7.39 (d,1H, J =8.4 Hz); 13C NMR (DMSO-d6) δ ppm: 22.6, 104.0, 114.3, 120.7, 132.8, 139.5, 145.6, 153.0, 168.6; LC-MS (ESI) m/z : 206.27 (M-H)–.
Procedure for synthesis N-(2-acetamido-1,3-benzothiazol-6-yl)-2-(1H-indol-3-yl)acetamide (3a)
To a solution of compound 2 (0.4g,.0019mol) in 12 mL THF ,indole-3-aceticacid(0.45g,0.0023),EDC.HCl(0.489g,0.0025m0l)and HOBt(0.260,0.0019 mol) was added. The reaction mixture was cooled to 00C and triethylamine was added then reaction mixture was allowed to RT and stirred for 8 hr.the reaction was monitored by TLC. The reaction solution was concentred under reduced pressure and partianated between ethylacetate (50mL) and water(40mL). The combined ethylacetate layer was washed with 2N HCl(1X25),10%NaHCO3(1X25,Brine(1x250mL) and dried over anhydrous Na2SO4. the resulting ethylacetate layer was washed and concentred under reduced pressure to afford compound 3a. Orange solid; yield 40%; m.p.254-256 oC; IR (KBr) νmax in cm-1: 3358 (NH), 1673 (C=O), 1575 (NH bend), 1273 (C-N), 731 (CH); 1H NMR (DMSO-d6) δ ppm: 2.18 (s,1H), 3.76 (s, 2H), 6.98 (t, 1H J =7.5 Hz), 7.07 (t,1H, J =7.5 Hz), 7.29 (d, 1H, J =1.5 Hz), 7.36 (d, 1H, J =8.1 Hz),7.55 (dd, 1H, J =1.5, 8.7 Hz),7.63 (d, 1H, J =7.5 Hz), 7.65 (d, 1H, J =8.7 Hz), 8.30 (d, 1H, J =1.5 Hz), 10.27 (s, 1H), 10.93 (s, 1H), 12.28(s, 1H); 13C NMR (DMSO-d6) δ ppm: 22.6, 33.7, 108.4, 111.2, 111.3, 118.3, 118.4, 120.3, 120.9, 123.7, 127.13, 131.7, 135.2, 136.0, 144.3, 156.7, 169.1,169.5; LC-MS (ESI) m/z : 365.27 (M+H)+; Anal. calcd. for C19H16N4O2S (364.42) %: C, 62.62; H,4.43; N, 15.37; Found: C, 62.72; H, 4.48; N, 15.22.
Procedure for synthesis N-(2-acetamido-1,3-benzothiazol-6-yl)-3-methoxybenzamide (3b)
Prepared as reported above for 3a starting from compound 2 and 3-methoxybenzoic acid. This reaction was carried out at room temperature for 6hr. Paleyellow solid; Yield 20%; m.p.257-258 oC; IR (KBr) νmax in cm-1: 3347 (NH), 3074, 2984 (CH), 1698 (C=O), 1621 (C=N), 1575 (NH bend), 1322 (C-N), 1273, 1039 (C-O), 710 (CH); 1H NMR (DMSO-d6) δ ppm: 2.19 (s, 3H), 3.84 (s, 3H), 7.16 (dd, 1H, J =2.1,7.8 Hz), 7.45 (t, 1H, J =7.8 Hz), 7.51 (d,1H, J =2.1 Hz),7.56 (d,1H, J =7.5 Hz), 7.74 (d, 1H, J =7.8 Hz), 7.70(d, 1H J =7.8 Hz), 8.41 (d, 1H), 10.36 (s, 1H), 12.30(s, 1H); 13C NMR (DMSO-d6) δ ppm: 22.7, 55.3, 112.8, 112.9, 117.2, 199.8, 119.9, 120.2, 129.5, 131.6, 134.9, 136.2, 144.8, 157.1, 159.1, 165.1, 169.2. LC-MS (ESI) m/z : 340.60 (M-H)–; Anal. calcd. for C17H15N3O3S (341.38) %: C, 59.81; H,4.43; N, 12.31; Found: C, 59.66; H, 4.49; N, 12.11.
Procedure for synthesis N-(2-acetamido-1,3-benzothiazol-6-yl)-2-(3-fluorophenyl)acetamide (3c)
Prepared as reported above for 3a starting from compound 2 and 3-fluorophenylacetic acid. This reaction was carried out at room temperature for 10 hr. Light brown solid; Yield 21%; m.p.274-275 oC;IR (KBr) νmax in cm-1: 3324 (NH), 3256, 3073, 3006 (CH), 1658 (C=O), 1565 (NH bend), 1235(C-F), 1283 (C-N), 754 (CH); 1H NMR (DMSO-d6) δ ppm: 2.18 (s, 3H), 3.75 (s, 2H), 8.27 (d, 1H, J =1.8 Hz), 7.66 (d, 1H, J =8.7 Hz), 7.52 (dd, 1H, J =1.8, 9 Hz), 7.40 (t, 1H, J =7.8 Hz), 7.31 (s, 1H), 7.18 (d, 1H, J =7.2 Hz),8.27(s, 1H), 10.37 (s, 1H), 12.28 (s, 1H); 13C NMR (DMSO-d6) δ ppm: 22.6, 36.16, 111.4, 115.0, 118.4, 120.3, 122.7, 122.9, 124.1, 124.2, 128.6, 128.7, 131.8, 134.9,144.4, 156.8, 167.8, 169.1; LC-MS (ESI) m/z : 344.20 (M+H)+; Anal. calcd. for C17H14 FN3O2S (343.08) %: C, 59.46; H, 4.11; N, 12.24; Found: C, 59.72; H, 4.05; N, 12.27.
Procedure for synthesis N-(2-acetamido-1,3-benzothiazol-6-yl)cyclopentanecarboxamide (3f)
Prepared as reported above for 3a starting from compound 2 and cyclopentane carboxylic acid This reaction was carried out at room temperature for 7 hr. White solid; Yield 27%;m.p.281-282 oC; IR(KBr) νmaxin cm-1: 3361 (NH), 3082, 2951, 2867 (CH), 1688 (C=O), 1610 (C=N), 1523 (NH bend), 1320 (C-N), 1464, 734 (CH bend); 1H NMR (DMSO-d6) δ ppm: 1.56-1.86 (m, 8H), 2.18 (s, 1H), 2.76-2.81 (m, 1H), 7.51 (d, 1H, J =8.4 Hz), 7.63(d, 1H, J =8.7 Hz),8.30 (s, 1H), 10.0 (s, 1H), 12.2 (s, 1H); 13C NMR (DMSO-d6) δ ppm: 22.3, 25.2, 29.7, 44.8, 110.9, 118.1, 119.9, 131.4, 135.0, 143.8, 156.33, 168.7, 173.8; LC-MS (ESI) m/z : 304.27 (M+H)+; Anal. calcd. for C15H17N3O2S (303.01) %: C, 59.38; H, 5.65; N, 13.85; Found: C, 59.36; H, 5.23; N, 13.82.
Procedure for synthesis N-(2-acetamido-1,3-benzothiazol-6-yl)-3,5-difluorobenzamide (3g)
Prepared as reported above for 3a starting from compound 2 and 3,5-difluorobenzoic acid, This reaction was carried out at room temperature for 6hr. Pale yellow solid; Yield 20%; m.p.282-283 oC; IR (KBr) νmax in cm-1: 3268 (NH), 3080 (CH), 1655 (C=O), 1592 (C=N), 1279 (C-N), 1123 (C-F),722 (CH bend); 1H NMR (DMSO-d6) δ ppm: 2.20 (s, 3H), 7.51-7.58 (m, 1H), 7.68 (s, 2H), 7.72 (d, 2H), 8.41(s, 1H), 10.51 (s, 1H), 12.33 (s, 1H); 13C NMR (DMSO-d6) δ ppm: 22.6, 110.8, 111.1, 133.0, 119.7, 120.2, 131.6, 134.3, 138.2, 145.1, 157.3, 160.4, 160.5, 162.6, 162.8, 169.2; LC-MS (ESI) m/z : 348.20 (M+H)+; Anal. calcd. for C16H11F2N3O2S (347.34) %: C, 55.33; H, 3.19; N, 12.10; Found: C, 55.32; H, 3.27; N, 12.54.
Procedure for synthesis N-(2-acetamido-1,3-benzothiazol-6-yl)-2-furamide (3d)
To the solution compound 2 (0.4g,0019 mol) in 12 mL THF,Triethylamine(0.8mL,0.0057mol) was added at 0oC. To the reaction mixtue 2-furoylchloride was added. The reaction mixture was allowed to RT and stirred for 4hr. The reaction was monitored by TLC. The reaction solution was concentrated under reduced pressure and partianated between ethylacetate (50mL) and water (40mL). The combined ethylacetate layer was washed with 2N HCl (1X30), 10%NaHCO3 (1X40, Brine (1x250mL) and dried over anhydrous Na2SO4. The resulting ethylacetate layer was washed concentrated under reduced pressure to afford compound 3d. Pale yellow solid; Yield 28%; m.p.287-289 oC; IR (KBr) νmax in cm-1: 3347 (NH), 3207, 3087 (CH), 1644 (C=O), 1603 (C=N), 1530 (NH bend), 1275 (C-N), 1163 (C-O), 734 (CH); 1H NMR (DMSO-d6) δ ppm: 2.19 (s, 3H), 6.71 (dd, 1H J =1.5, 3.3 Hz), 7.35 (1H, d, J =3.3Hz), 7.69 (d,1H, J=8.7 Hz), 7.73 (d, 1H, J =8.7Hz), 7.95 (s,1H, J=1.5 Hz), 10.34 (s, 1H),12.31 (s, 1H); 13C NMR (DMSO-d6) δ ppm: 22.6, 112.0, 112.8, 114.6, 119.7, 120.2, 131.6, 134.2, 144.8, 145.6, 147.4, 156.0, 157.0, 169.1; LC-MS (ESI) m/z : 302.20 (M+H)+; Anal. calcd. for C14H11N3O3S (301.32) %: C, 55.80; H, 3.68; N, 13.95; Found: C, 55.68; H, 3.45; N, 13.27.
procedure for synthesis (2E)-N-(2-acetamido-1,3-benzothiazol-6-yl)-3-(2-furyl)acrylamide (3e)
Prepared as reported above for 3g starting from compound 2 and (2E)-3-(furan-2-yl)acryloyl chloride . This reaction was carried out at room temperature for 3hr. yellow solid;yield 35%; m.p.257-258 oC; IR (KBr) νmax in cm-1: 3351 (NH), 1669 (C=O), 1605 (C=N), 1469 (CH bend), 1273 (C-N), 1173 (C-O), 748(CH); 1H NMR (DMSO-d6) δ ppm: 2.19 (s, 3H), 6.62 (d, 1H, J =3.6 Hz), 6.65 (1H, d, J =15.5 Hz), 6.86 (d, 1H J =3.3 Hz), 7.40 (d, 1H, J =15.3 Hz), 7.56 (dd, 1H, J =1.5, 8.7 Hz), 7.68 (d,1H, J =8.7 Hz), 7.83 (s, 1H), 8.42 (d, 1H, J =1.5 Hz), 10.37 (s, 1H), 12.29 (s, 1H); 13C NMR (DMSO-d6) δ ppm: 22.7, 111.4, 112.5, 114.5, 118.4, 119.3, 120.5, 127.2, 131.9, 135.2, 144.5, 145.5, 145.1, 150.9, 156.9, 163.2, 169.2; LC-MS (ESI) m/z : 328.27 (M+H)+; Anal. calcd. for C16H13N3O3S (327.36) %: C, 58.70; H, 4.00; N, 12.84; Found: C, 58.88; H, 3.97; N, 12.90.
procedure for synthesis N-(6-{[(4-tert-butylphenyl)sulfonyl]amino}-1,3-benzothiazol-2-yl)acetamide(4a)
To the solution compound 2 (0.4g,0019 mol) in 10 mL 1,2-dichloroethane,pyridine (0.46mL,0.0057mol) and 4-tert-butylbenzene-1-sulfonylchloride(0.45g,0.0019 mol) was added. The reaction mixture was heated to 90oC for 7 hr. reaction was monitored by TLC. The reaction mixture was cooled to RT ,diluted with 30 mL of ethyl acetate. The combined ethylacetate layer was washed with 2N HCl(1X30),10%NaHCO3(1X40,Brine(1x50mL) and dried over anhydrous Na2SO4.the resulting ethylacetate layer was concentrated under reduced pressure to afford compound 4a.white solid;yield 39%; m.p.265-266 oC; IR (KBr) νmax in cm-1: 3310 (NH), 2961(CH), 1690 (C=O), 1606 (C=N), 1546 (NH bend), 1275 (C-N), 1328, 1158 (S=O), 734 (CH bend); 1H NMR (DMSO-d6) δ ppm: 1.23 (s, 9H), 2.17 (s, 3H), 7.16 (dd, 1H, J =2.1, 8.7 Hz), 7.57 (d, 1H, J =8.7 Hz), 7.54 (d, 2H, J =8.4 Hz), 7.69 (d, 2H J =8.4 Hz), 7.66 (s, 1H), 10.32 (s, 1H), 12.29 (s, 1H); 13C NMR (DMSO-d6) δ ppm: 20.6, 30.6, 34.7, 113.0, 119.6, 120.7, 125.9, 126.4, 132.1, 133.4, 136.6, 145.2, 155.7, 157.3, 169.2; LC-MS (ESI) m/z : 404.20 (M+H)+; Anal. calcd. for C19H21N3O3S2 (403.52) %: C, 56.55; H, 5.25; N, 10.41; Found: C, 56.63; H, 5.79; N, 10.32.
Procedure for synthesis N-(6-{[(4-cyclohexylphenyl)sulfonyl]amino}-1,3-benzothiazol-2-yl)acetamide (4b)
Prepared as reported above for 4a starting from compound 2 and 4-cyclohexylbenzene-1-sulfonylchloride. This reaction was carried out at 90oC for 10hr. White solid; Yield 39%; m.p.267-268oC; IR (KBr) νmax in cm-1: 3312 (NH), 2924, 2852 (CH), 1689 (C=O), 1605 (C=N), 1549 (NH bend), 1470 (CH bend), 1275 (C-N), 1327, 1154 (S=O); 1H NMR (DMSO-d6) δ ppm: 1.29-1.32 (m, 5H), 1.64-1.73 (m, 5H), 2.16 (s, 3H), 2.51 (m, 1H), 7.13 (dd, 1H, J =2.1, 8.7 Hz), 7.36 (d, 2H J =8.4 Hz), 7.57 (d, 1H, J =8.7 Hz), 7.64 (s, 1H), 7.66 (d, 2H J =8.4 Hz), 10.28 (s,1H), 12.28 (s,1H); 13C NMR (DMSO-d6) δ ppm: 22.6, 25.3, 26.0, 33.3, 43.4, 113.2, 119.8, 120.7, 126.7, 127.4, 132.1, 133.4, 136.9, 145.3, 152.6, 157.4, 169.3; LC-MS (ESI) m/z : 430.27 (M+H)+; Anal. calcd. for C21H23N3O3S2 (429.56) %: C, 58.72; H, 5.40; N, 9.78; Found: C, 58.45; H, 5.92; N, 9.68.
Procedure for synthesis N-(6-{[(2,5-dichloro-3-thienyl)sulfonyl]amino}-1,3-benzothiazol-2-yl)acetamide (4c)
Prepared as reported above for 4a starting from compound 2 and 2,5-Dichlorothiophene-3-sulfonyl chloride. This reaction was carried out at 90oC for 8 hr. White solid; Yield 30%; m.p.262-263 oC; IR (KBr) νmax in cm-1: 3410 (NH), 3062, 2973(CH), 1675 (C=O), 1561 (NH bend), 1276(C-N), 1345, 1163 (S=O), 1044(C-Cl), 725(CH bend); 1H NMR (DMSO-d6) δ ppm: 2.18 (s, 3H), 7.16 (dd, 1H, J =2.1,8.7 Hz), 7.31 (s, 1H), 7.64 (d, 1H, J =8.7 Hz), 7.72 (d, 1H, J =2.1 Hz), 10.70 (s, 1H), 12.35 (s, 1H); 13C NMR (DMSO-d6) δ ppm: 22.6, 114.4, 120.5, 120.8, 126.3, 126.7, 129.5, 131.9, 132.1, 135.4, 145.9, 157.8, 169.3; LC-MS (ESI) m/z : 423.87 (M+H)+; Anal. calcd. for C13H9Cl2N3O3S3 (422.33) %: C, 36.97; H, 2.15; N, 9.95; Found: C, 36.92; H, 2.23; N, 9.92.
Procedure for synthesis N-(6-{[(4-fluorophenyl)carbamoyl]amino}-1,3-benzothiazol-2-yl)acetamide (5a)
To the solution compound 2 (0.3g,0015 mol) in 6 mL THF, 4-fluorophenylisocyanate (0.24g,0.0017mol) in 3 mL dichloromethane was added at 0oC. The reaction mixture was allowed to RT and stirred for 1 hr.The reaction mixture was concentrated under reduced pressure and partianated between ethyl acetate (50mL) and wate r(40mL). The ethylacetate layer was washed with Brine(1x250mL) and dried over anhydrous Na2SO4. The resulting ethylacetate layer was washed concentrated under reduced pressure. The residue formed was washed with 20mL of diethyl ether and dried under vacuum to afford 5a. Light brown solid; Yield 25%; m.p.262-263 oC; IR (KBr) νmax in cm-1: 3278 (NH), 3068, 2997 (CH), 1635 (C=O), 1557 (NH bend), 1216 (C-F), 1270 (C-N), 719 (CH bend); 1H NMR (DMSO-d6) δ ppm: 2.21(s, 3H), 7.14 (t, 2H, J=9Hz), 7.44 (dd, 1H, J =3,9Hz), 7.49 (1H, d , J =9 Hz), 7.52 (1H, d, J =9Hz), 8.15 (d, 1H, J =3Hz), 8.77 (s, 1H), 8.83 (s, 1H), 12.27 (s, 1H); 13C NMR (DMSO-d6) δ ppm: 23.1, 110.9, 115.7, 118.4, 120.4, 120.9, 132.6, 136.9, 136.50, 144.18, 153.2, 156.5, 159.3, 169.6; LC-MS (ESI) m/z : 345.27 (M+H)+; Anal. calcd. for C16H13FN4O2S (344.36) %: C, 55.80; H, 3.81; N, 16.27; Found: C, 55.72; H, 3.77; N, 16.54.
Procedure for synthesis N-(6-{[(4-fluorophenyl)carbamothioyl]amino}-1,3-benzothiazol-2-yl)acetamide (5b)
Prepared as reported above for 5a starting from compound 2 and 4-fluorophenylisothiocyanate. This reaction was carried out at RT for 1 hr. White solid; Yield 36%; m.p.231-232 oC; IR (KBr) νmax in cm-1: 3330 (NH), 2931 (CH), 1688 (C=O), 1600 (C=N), 1552 (NH bend), 1265 (C-N), 1220 (C-F), 723(CH bend); 1H NMR (DMSO-d6) δ ppm: 2.21 (s, 3H), 7.17 (t, 2H, J =9Hz), 7.43 (d,1H J =9Hz), 7.48 (d, 2H, J =9Hz), 7.69 (d, 1H, J =9Hz), 8.03 (s, 1H), 9.75 (s, 1H), 9.90 (s,1H); 13C NMR (DMSO-d6) δ ppm: 27.9, 120.3, 122.5, 125.3, 128.9, 131.6, 136.7, 140.1, 141.0, 151.1, 162.8, 162.9, 174.5, 185.5; LC-MS (ESI) m/z : 361.27 (M+H)+; Anal. calcd. for C16H13FN4OS2(360.43) %: C, 53.32; H, 3.64; N, 15.54; Found: C, 53.38; H, 3.48; N, 15.77.
Pharmacology
Cell lines
The human breast adenocarcinoma (MCF 7), human cervical adenocarcinoma (HeLa) and human osteosarcoma (MG63) cell lines were obtained from National Centre for Cell Science (NCCS), Pune and grown in Minimum Essential Medium containing 10% fetal bovine serum (FBS). The cells were maintained at 370C, 5% CO2, 95% air and 100% relative humidity. Maintenance cultures were passaged weekly, and the culture medium was changed twice a week.
In vitro cytotoxic activities (MTT assay)
The monolayer cells were detached with trypsin-ethylenediaminetetraacetic acid (EDTA) to make single cell suspension and viable cells were counted using a hemocytometer and diluted with medium containing 5% FBS to give final density of 1×105 cells/ml. One hundred microlitres per well of cell suspension were seeded into 96-well plates at plating density of 10,000 cells/well and incubated to allow for cell attachment at 370C, 5% CO2, 95% air and 100% relative humidity. After 24 hr, the cells were treated with serial concentrations of the test samples. They were initially dissolved DMSO, and an aliquot of the sample solution was diluted to twice the desired final maximum test concentration with serum-free medium. Additional four serial dilutions were made to provide a total of five sample concentrations. Aliquots of 100 µl of these different sample dilutions were added to the appropriate wells already containing 100 µl of medium, resulting in the required final sample concentrations. Following sample addition, the plates were incubated for an additional 48 h at 370C, 5% CO2, 95% air, and 100% relative humidity. The medium containing without samples were served as control and triplicate were maintained for all the concentrations. After 48 h of incubation, 15 µl of MTT (5mg/ml) solution was added to each well and incubated at 370C for 4 h. The medium with MTT was then flicked off, and the formed formazan crystals were solubilized in 100 µl of DMSO and then measured the absorbance at 570 nm using a micro plate reader. The % cell inhibition was determined using the following formula.
% Cell Inhibition = 100- Abs (sample)/Abs (control) x100.
Results and Discussion
Chemistry
The route for the synthesis of the intermediates and target compounds are shown in the scheme (1-3). The Intermediate 1 was prepared by using the reagent acetic anhydride and pyridine. The FT-IR value at 1701 cm-1(for C=O), 1H NMR value δ 2.23 (3H, singlet) and 13C NMR δ 170.1(C=O) which confirms that there is the formation of the acetylated product. The intermediate 2 was synthesized by using the reagent stannous chloride and Con.HCl. The FT-IR value at 3417,3298(for NH2) and 1H NMR δ 5.13 (2H, broad peak) which confirm that there is the formation of reduction product. The resulting primary amine also confirmed by ninhydrin activity in TLC. The compound 2 was taken as a common scaffold for the synthesis of the new series of 2, 6-disubstituted benzothiazole derivatives.
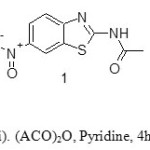 |
Scheme1: Reagents and conditions: (i). (ACO)2O, Pyridine, 4h,90oC; (ii). SnCl2 .2H2O, HCl,2h, RT. Click here to View scheme |
First the amide derivatives 3a,3b,3c,3f,3g was prepared by using the reagent 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide(EDCI).The corresponding acid chloride was used for making of amide derivatives for 3d,3e. For all the amide derivatives, the two amide NH observed at δ 10-10.5 and δ 12-12.5 as a broad singlet in 1H NMR.
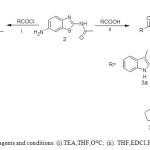 |
Scheme2: Reagents and conditions: (i).TEA,THF,OOC; (ii). THF,EDCI,HOBt,TEA. Click here to View scheme |
The sulfonamide derivatives 4a, 4b, 4c was prepared by using corresponding sulfonyl chloride and compound 2. The reaction was carried out in the presence of pyridine base at 90oC.The FT-IR stretching frequency 1320-1350 cm-1 and 1150-170 cm-1 (S=O) conform that there is the formation of sulfonamide derivatives. The sulfonamide NH chemical shift value in 1H NMR observed at δ10-10.5 as a broad singlet. The corresponding isocyanate and isothiocyanate respectively was used to prepare urea and thiourea derivative 5a and 5b.
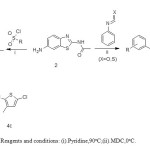 |
Scheme3: Reagents and conditions: (i).Pyridine,90oC;(ii).MDC,0oC. Click here to View scheme |
Biology
Cytotoxicity
In vitro cytotoxicity of the synthesized compounds were assessed by standard 3-(4, 5-imethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in MCF-7, HeLa, and MG63 cell lines. Among the synthesized compounds 3a, 3f, 3g, and 4b showed cytotoxic effects on all the three cell lines after 48 h exposure. Maximum cytotoxicity was obtained for the sulfonamide derivative 4b in all the three cell lines when compared to others, and the IC50 was 36, 44 and 34 µM against MCF7, HeLa and MG63 respectively. The amide derivative 3a also showed cytotoxicity in all the three cell lines and the IC50 was in the range of 48 to 53 µM. The remaining urea and thiourea derivatives did not show any cytotoxicity activity at the tested concentrations. For standard cisplatin was used. All the results are showed in Table 1.
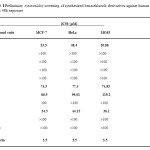 |
Table1: Preliminary cytotoxicity screening of synthesized benzothiazole derivatives against human cancer cell lines at 48h exposure Click here to View table |
The known numbers of cells (1×105 cells/ml) were incubated for 24 h in a 5% CO2 Incubator at 37oC in the presence of different concentrations of test compounds. The medium with MTT was then flicked off, and the formed formazan crystals were solubilized in 100 µl of DMSO and then measured the absorbance at 570 nm using a micro plate reader.
Conclusions
A series of new 2,6-disubstituedbenzothiazol derivatives were synthesized, and their anti-cancer activities were evaluated in vitro. The results showed that the cyclohexyl benzene sulphonamide derivative of benzothiazole(4b) showed good activity against MCF-7, HeLa, and MG63 cells. Further amide derivatives 3a,3g,3h also show good activity against MCF7, HeLa, and MG63. These data suggested that the compound 4b may be powerful anticancer agent and be worth being further investigated as a potential of an anticancer agent.
Acknowledgement
The authors are very much thankful to Dr.R.S.D. Wahida Banu, Principal, Government College of Engineering, Salem-636 011, Tamil Nadu for providing the facilities to carry out this research work and for her constant encouragement.
References
- Reddy, P.; Lin, Y.; Chang, H. Synthesis of novel benzothiazole compounds with an extended conjugated system. Arcivoc. 2007; xvi: 113-122.
- Piscitelli, F.; Ballatore, C.; Smith, A. Solid Phase synthesis of 2- aminobenzothiazoles. Bioorg.Med Chem Lett.2010; (20):644-648.
- Shi, D.; Bradshaw, T. D.; Wrigley, S.; McCall, C. J.; Lelieveld, P.; Fichtner, I.; Stevens, M.F G. J. Med. Chem.1996, 37, 3375.
- Bradshaw, T. D.; Shi, D. F.; Schultz, R. J.; Paull, K. D.; Kelland. L.; Wilson, A.; Garner, C.; Fiebig, H. H.; Wrigley, S.; Stevens, M. F. G. Br. J. Cancer 1998, 77, 421.
- Kashiyama, E.; Hutchinson, I.; Chua, M.S.; Stinson, S. F.; Phillips, L. R.; Kuar, G.;Sausville, E. A.; Bradshaw, T. D.; Westwell, A. D.; Stevens, M. F. G. J. Med. Chem. 1999,42, 4172.
- Quiroga, J.; Hernandez, P.; Insuasty, B.; Abonıa, R.; Cobo, J.; Sanchez, A.; Nogueras, M.; Low, J.N. Control of the reaction between 2-aminobenzothiazoles and Mannich bases.Synthesis of pyrido[2,1- b][1,3]benzothiazoles versus [1,3]benzothiazolo[2,3-b]quinazolines.J. Chem. Soc.Perkin Trans. I 2002, 1, 555–559.
- Kok, S.H.L.; Chui, C.H.; Lam, W.S.; Chen, J.; Lau, F.Y.; Wong, R.S.M.; Cheng, G.Y.M.; Lai, P.B.S.; Leung,R.W.T.; Tang, J.C.O.; et al. Synthesis and structure evaluation of a Novel cantharimide and its cytotoxicity on SK-Hep-1 hepatoma cells. Bioorg. Med. Chem. Lett.2007,17, 1155–1159.
- Kok, S.H.L.; Chui, C.H.; Lam, W.S.; Chen, J.; Lau, F.Y.; Wong, R.S.M.; Cheng, G.Y.M.; Tang, W.K.; Teo, I.T.N.; Cheung, F.; et al. Apoptogenic activity of a synthetic cantharimide in leukaemia: Implication on its structural activity relationship. Int. J. Mol. Med. 2006, 18, 1217–1221.
- Xuan-Hong, Shi.; Zhao, Wang.; Yong Xia, Ting-Hong, Ye.; Mei, Deng.; You-Zhi, Xu.; Yu- Quan, Wei and Luo- Ting Yu .Synthesis and Biological Evaluation of Novel Benzothiazole-2-thiol Derivatives as Potential Anticancer Agents Molecules 2012, 17,3933-3944.
- Sohail Saeed, a.; Naghmana Rashid, a.; Peter, G.; Jones, b.; Muhammad Ali, c.;, Rizwan Hussain Synthesis, characterization and biological evaluation of some thiourea derivatives bearing benzothiazole Moiety as potential antimicrobial and anticancer agents, European Journal of Medicinal Chemistry 45 (2010)1323–31.
- Dao Thi Kim Oanh, a.; Hoang Van Hai, a.; Sang Ho Park, b.; Hyun-Jung Kim, c.; Byung-Woo Han, b.;Hyung- Sook Kim, d.; Jin-Tae Hong, d.; Sang-Bae Han, d.; Van Thi My Hue, a.;Nguyen-Hai Nama,Benzothiazole-containing hydroxamic acids as histone deacetylase inhibitors and antitumor agents, Bioorganic & Medicinal Chemistry Letters 21 2011 7509–7512.
- Mortimer, C.; Wells, G.; Crochard, J.P .; Stone, E.L.; Bradshaw, T.D.; Stevens, M.G.F.; West well, A.D. J. Med. Chem. 49 (2006) 179–185.
- Yoshida, M.; Hayakawa, I.; Hayashi, N.; Agatsuma, T.; Oda, Y.; Tanzawa, F.; Iwasaki, S.; Koyama, K.; Furukawa, H.; Kurakata, Y.; Sugano, Y. Bioorg. Med. Chem. Lett. 15 (2005) 3328–3332.
- Vicini, P.; Gernonikaki, A.; Incerti, M.; Busonera, B.; Poni, G.; Cabras, C.A.; Colla, P.L. Bioorg. Med. Chem.11 (2003) 4785–4789.
- Pattan, S.R,; Narendra babu, S.N. Synthesis and antifungal activity of 2-amino [5’-(4-sulphonyl benzylidene)-2, 4- thiazolidene Dione] -7- (substituted)-6-fluro benzothiazoles, Ind J of Het. Chem., 2002, 11,333-334.
- Huang, S.T.; IJen, H.; Synthesis and anticancer evaluation of bis (benzimidazoles), bis (benzoxazoles), and benzothiazoles, Bio. and Med. Chem., 2006, 146, 106-6119.
- Kok, S.H.L.; Gambari, R.; Chui, C.H.; Yuen, M.C.W.; Lin, E.; Wong, R.S.M.; Lau, F.Y.; Cheng, G.Y.M.; Lam, W.S.; Chan, S.H.; et al. Synthesis and anti-cancer activity of benzothiazole containing phthalimide on human carcinoma cell lines. Bioorg. Med. Chem. 2008, 16, 3626–3631.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


