Synthesis and Antioxidant Ability of 5-amino-1,3,4-oxadiazole Derivitives Containing 2,6-dimethoxyphenol
Khalid Fahad Ali*
Department of Chemistry, Ibn Al-haitham, University of Baghdad, Baghdad 61023, Iraq
DOI : http://dx.doi.org/10.13005/ojc/310126
Article Received on :
Article Accepted on :
Article Published : 17 Feb 2015
4-(((4-hydroxy-3,5-dimethoxybenzyl)oxy)methyl)benzoic acid was synthesized from multisteps and converted to their corresponding hydrazide. The corresponding hydrazide was cyclized to their corresponding 5-amino-1,3,4-oxadizole. Newly Schiff bases (7a-7e) were synthesized from reaction the 5-amino-1,3,4-oxadizole with several substituted of 4-hydroxybenzylaldehyde. The resulting compounds were characterized based on their IR, 1H-NMR, 13C-NMR, and HRMS data. 2,2-Diphenyl-1-picrylhydrazide (DPPH) and ferric reducing antioxidant power (FRAP) assays were used to test the antioxidant properties of the synthesized compounds. Compound 7d and 7e exhibited significant free-radical scavenging ability in both assays.
KEYWORDS:5-amino-1; 3; 4-oxadiazole; Schiff base; antioxidant; DPPH; FRAP
Download this article as:| Copy the following to cite this article: Ali K. F. Studies on Chemical, Synthesis and Antioxidant Ability of 5-amino-1,3,4-oxadiazole Derivitives Containing 2,6-dimethoxyphenol. Orient J Chem 2015;31(1). |
| Copy the following to cite this URL: Ali K. F. Studies on Chemical, Synthesis and Antioxidant Ability of 5-amino-1,3,4-oxadiazole Derivitives Containing 2,6-dimethoxyphenol. Orient J Chem 2015;31(1). Orient J Chem 2015;31(1). Available from: http://www.orientjchem.org/?p=7260 |
Introduction
The reactive oxygen species (ROS) and other related free radicals species are capable to react either directly or indirectly, to damage all biomolecules, such as proteins, lipids, DNA and carbohydrates resulting in cell damage and subsequently this damage can cause many diseases 1. Many researchers reported the implication of these free radicals will cause several diseases such as inflammatory2, cancers3, degenerative4 and chronic diseases5. Phenolic antioxidant compounds are one of the most important antioxidant species which can inhibit the oxidative stress in biological system and prevent any damage. These compounds also show broad biological activity as anti inflammatory6 and anticancer7.
Generally, antioxidants donate protons to become stable free radicals. This stability increases with the extent of delocalization and enhances antioxidant ability8,9 . As such, many synthesized compounds containing long chain resonance exhibited high antioxidant activity. Furthermore, the compounds which can be classified as a strong antioxidant usually shared common structure features. They often own multiple phenolic hydroxyl groups like flavonoids 10 or which have full conjugation π system like carotenoids11. Moreover, exhibited substituted groups might influence on the scavenging ability. This indicates the existence of a close relationship between the chemical structure and the ability to scavenge free radicals.
Derivatives of 1,3,4-oxadiazole are shown to exhibit various types of biological activities 12-14and antioxidant ability 9,15,16and compounds of Schiff base derivatives have even broader properties and applications such as liquid crystalline 17,18, nonlinear optical devices 19, dyes 20, biological 21-23 and pharmaceutical 24 applications. As for 2,6-dimethoxyphenol derivatives, there has been widespread interest during the last years as antioxidant materials25,26 ..n this work we presented the synthesis of new 4-(((4-(5-((aryidene)amino)-1,3,4-oxadiazol-2-yl)benzyl)oxy) methyl)-2,6-dimethoxyphenol (7a-7e) as new antioxidant material.
Material and Method
Chemistry
The IR spectra were obtained with a Perkin Elmer 400 Fourier Transform Infrared (FTIR) Spectrometer. 1H and 13C-NMR spectra were recorded at Joel Lambda and ECA DELTA spectrometers at 400 MHz) UM, Malaysia). CDCl3 and DMSO-d6 were used as solvents with TMS as the internal standard. The mass spectra were recorded using an Agilent 5975 system for EI/MS and a Finnigan TSQ7000 for HREIMs (NUS, Singapore). For UV spectroscopy, a Power Wave X 340 (BIO-TEK Instruments, Inc., Winooski, VT, USA) was used to record the FRAP and DPPH. Melting points were measured on a Gallenkamp melting point apparatus in open-end capillary tubes. Flash column chromatography on silica gel 60 (230–400 mesh, E. Merck) was employed. General grade solvents and reagents were purchased from commercial suppliers and used without further purification.
Synthesis of 3,5-dimethyl-4-((trimethylsilyl)oxy)benzaldehyde (1)
4-Hydroxy-3,5-dimethylbenzaldehyde (3.64 g, 20 mmol) was dissolved in dry DMF (15 mL) containing anhydrous potassium carbonate (2.76 g, 25 mmol) . The mixture stirred for 30 minutes at ambient temperature, then Chlorotrimethylsilane (3.25, 30 mmol) was added drop wise. The stirring mixture was heated under reflux (70-80 ̊C) for 24 h. After cooling the resulting product was extracted from diethyl ether (3 x 50 mL) and dried under calcium chloride. The crude product was purified by column chromatography using hexane-ethylacitate (8:2) as eluent to obtain colorless oil, Yield; 74%, Bp, 288-292 ̊C at 760 mmHg, d=1. 23 at 25 ̊C. IR (liquid film) vmax 3033(CHAr), 2973, 2958(CHaliphatic) ,1674(CO), 1595(C=C), 1190 (Ar-O-C), 842(Si-CH3) cm-1, 1H-NMR(400MHz, CDCl3): δ 0.23(s, 9H, Si-(CH3)3), 3.84(s, 6H, 2×OCH3)7.13(s, 2H, H-3), 9.85(s, 1H, CHO). 13C-NMR (100 MHz, CDCl3): δ -0.08 (3C, Si-(CH3)3, 55.42(2C, OCH3), 108(2C, C-3), 131.63(1C,C-4), 134.17(1C, C-3), 151.83(1C, C-2), 192.07(1C, CO). HREIMs m/z = 254.0968 [M˙+] (calc. for C12H18O4Si, 254.0974).
Synthesis of 3,5-dimethyl-4-((trimethylsilyl)oxy)benzyl alcohol (2)
Sodium borohydride (0.75 gm, 20 mmol) was added with small portion to a stirred solution of 3,5-dimethyl-4-((trimethylsilyl)oxy)benzaldehyde (3 g, 13.3 mmol) in THF-Methanol 4:1 (25 mL) and refluxed overnight. Upon cooling distill water (50 mL) was added and the mixture was stirred for further 10 minutes. The crude product was extracted from diethyl ether. The organic layer washed three times with saturated solution of Sodium hydrogen sulfate. After evaporating the solvent pale yellow oil was obtained. Yield 87%, Bp 314-317 ̊C at 760 mm Hg, d=1.12 at 25 ̊C. IR (liquid film) vmax 3329(OH), 3067(CHAr), 2943, 2877 (CHaliphatic), 1593(C=C), 1205 (Ar-O-C), 859(Si-CH3) cm-1, 1H-NMR(400 MHz, CDCl3): δ 0.20(s, 9H, Si-(CH3)3), 2.94 (bs, 1H, OH), 3.82 (s, 6H, 2×OCH3), 4.51(s, 2H, OCH2), 6.67(s, 2H, H-3). 13C-NMR (100 MHz, CDCl3): δ -0.05 (3C, Si-(CH3)3, 55.42(2C, OCH3), 62.34(1C, CH2OH), 107.52(2C, C-3), 128.44(1C, C-1), 135.68(1C, C-4), 152.08(1C, C-2), HREIMs m/z = 256.1129 [M˙+] (calc. for C12H20O4Si, 256.1131).
Synthesis of 4-(((3,5-dimethoxy-4-((trimethylsilyl)oxy)benzyl)oxy) methyl)benzonitrile (3)
Small portions of 4-(bromomethyl)benzonitrile (2.3g, 11.6 mmol) was added to a stirring solution of 3,5-dimethyl-4-((trimethylsilyl)oxy)benzyl alcohol (2.6 g,11.6 mmol) in pyridine (15 mL ) within 30 minutes. After complete the addition the mixture was refluxed for 6h. Upon cooling the mixture poured in to crashed water (100 mL) and acidified with 5% of hydrochloric acid. The product was extracted with diethyl ether (3×25 mL) and washed with water, then dried over magnesium sulfate. After evaporating the solvent, the crude material was purified by column chromatography using hexane-ethylacetate (9:1) as eluent to obtain colorless oil which is solidify after two days to obtain white powder. Yield 61%, Mp 36-38 ̊C, IR (KBr) vmax 3042 (CHAr), 2962, 2890 (CHaliphatic), 2248(C≡N),1595(C=C), 1195 (Ar-O-C), 867(Si-CH3) cm-1. 1H-NMR (400MHz, CDCl3): δ 0.18 (s, 9H, Si-(CH3)3), 3.89(s, 6H, 2×OCH3), 4.31 (s, 2H, OCH2), 4.35 (s, 2H, OCH2), 6.62 (s, 2H, H-3), 7.41 (d, 2H, J 8.2, H-8),7.49(d, 2H, J 7.9, H-9). 13C-NMR (100 MHz, CDCl3): δ -0.046 (3C, Si-(CH3)3, 56.02(2C, OCH3), 71.14(1C, CH2OCH2), 70.67(1C, CH2OCH2), 101.22(1C, CN ), 107.75(2C, C-3), 127.89(1C,C-1), 128.55(2C,C-8), 131.87(2C,C-9), 135.43(1C,C-10), 138.44(1C-C-4), 140.2(1C-C7) 151.98(2C, C2), HREIMs m/z = 318.1097 [M˙+] (calc. for C17H18O6, 318.1103).
Synthesis of 4-(((4-hydroxy-3,5-dimethoxybenzyl)oxy)methyl)benzoic acid (4)
4-(((3,5-dimethoxy-4-((trimethylsilyl)oxy)benzyl)oxy)methyl)benzonitrile (2.2 g, 6 mmol) was dissolved in ethanol (25 mL) and 5 mL of 4N KOH solution. The mixture was refluxed for 48 h. After evaporation the solvent the crude product extract with 20 mL ethyl acetate The organic layer was ignored. The aqueous layer reduced to 5mL by evaporation under reduced pressure. of acetic acid-THF 1:1 (10mL) was added to the aqueous layer then refluxed overnight. The precipitated collected by filtration and washed with water. Recrystallized with ethanol to obtain white microcrystals. Yield 66 %, Mp 152-154 ̊C, IR (KBr) vmax 3350 (OH), 3042 (CHAr), 2962, 2890 (CHaliphatic), 1673(C=O) 1595(C=C), 1195 (Ar-O-C), cm-1. 1H-NMR (400MHz, CDCl3): 3.89(s, 6H, 2×OCH3), 4.32 (s, 2H, OCH2), 4.37 (s, 2H, OCH2), 6.62 (s, 2H, H-3), 7.62 (d, 2H, J 8.2, H-8),8.22(d, 2H, J 8.0, H-9). 9.12 (bs, 1H, OH), 13C-NMR (100 MHz, CDCl3): 56.02(2C, OCH3), 71.14(1C, CH2OCH2), 70.67(1C, CH2OCH2), 108.35(2C, C-3), 128.71(2C,C-8), 129.26(1C,C-10), 130.43(2C, C-9), 133.32(1C, C-4), 139.44(1C, C-1), 142.08(1C, C-7) 151.98 (2C, C2), 169.6(1C, C=O) HREIMs m/z = 318.1100 [M˙+] (calc. for C17H18O6), 318.1103).
Synthesis of 4-(((4-hydroxy-3,5-dimethoxybenzyl)oxy)methyl) benzohydrazide (5)
Thionylchloride (4 mL) was added in small portions of 4-(((4-hydroxy-3,5-dimethoxybenzyl)oxy)methyl)benzoic acid to (2 g,6.2 mmol). The mixture was refluxed for 3h, the excess of thionyl chloride was removed under reduced pressure. 0.1 moles of acid chloride (without further purification) were dissolved in dry benzene(15 mL), and it was transferred to an addition funnel hydrazine hydrate (98%) 5 mL in dried benzene (10 mL) was added into a two neck flask that equipped with a condenser. The addition funnel was then fixed onto the flask and secured firmly. The acid chloride was added drop wise at 0 ̊C. After that, the mixture was allowed to stand for 1 h at an ambient temperature. It was then stirred and refluxed for 3 h.The excess solvent was removed under reduced pressure and the crude solid was collected, wash with water and recrystallized from ethanol afforded white solid. Yield 84%, Mp 92-94 ̊C , IR (KBr) vmax 3418.2 (OH phenol), 3298, 3210 (NH, NH2), 3055 (CHAr), 2958 -2880 (CHaliphatic), 1665 ( C=O), 1595 (C=C), 1197(Ar-O-C), cm-1. 1H-NMR (400MHz, DMSO-d6): 3.68(s, 6H, 2× OCH3), 4.33 (s, 2H, OCH2), 4.29 (s, 2H, OCH2), 4.74(bs, 2H, NH2) 6.56 (s, 2H, H-3), 7.69 (d, 2H, J 8.2, H-8), 8.22(d, 2H, J 7.88, H-9),8.83(bs, 1H, CONH), 9.14(bs, 1H, OH), 13C-NMR (100 MHz, DMSO-d6): 56.1(2C, OCH3), 71.11(1C, CH2OCH2), 70.59(1C, CH2OCH2), 107.85(2C, C-3), 129.01(2C,C-8), 128.96(1C, C-10), 130.13(2C, C-9), 133.33(1C, C-4), 138.84(1C, C-1), 141.78(1C, C-7) 152.08 (2C, C-2), 166.11(C=O). HREIMs m/z = 332.1368 [M˙+] (calc. for C17H20N2O5, 332.1372).
Synthesis of 4-(((4-(5-amino-1,3,4-oxadiazol-2-yl)benzyl)oxy)methyl)-2,6-dimeth oxyphenol (6)
A solution of hydrazide, 5 (2 g,6 mmol) in methanol (10 mL) and sodium bicarbonate (0.5, 6 mmol) was stirred in 50 mL round bottom flask, then cyanogene bromide ((0.66 gm, 6.3 mmol) was added. The mixture was left stirring at ambient temperature overnight. After that, cold water (5 mL) was added to the mixture and the precipitate was collected and dried at 60 °C. The white solid was recrystallized from ethanol to give (62%) yield; Mp 132-134 °C, IR (KBr) vmax 3468(br, OHphenol), 3318, 32 (NH2), 3093( CHAr), 2960, 2870 (CHaliphatic): 1609(C=N), 1600, 1552(C=C), 1250(C-N), 1203(C-O), 1099 (C-O-C); cm-1; 1H-NMR (400MHz, DMSO-d6)3.71(s, 6H, 2× OCH3), 4.42 (s, 2H, OCH2), 4.45 (s, 2H, OCH2), 6.57 (s, 2H, H-3), 7.51(bs, 2H, NH2) 7.72 (d, 2H, J 8.22, H-8), 8.18(d, 2H, J 8.2,H-9), 9.11(bs, 1H, OH), 13C-NMR (100 MHz, DMSO-d6): 56.12(2C, OCH3), 71.21(1C, CH2OCH2), 69.91(1C, CH2OCH2), 107.85(2C, C-3), 129.06(2C, C-9), 129.15(1C, C-10), 131.33(2C, C-8), 133.04(1C, C-4), 138.75(1C, C-1), 142.07 (1C, C-7) 151.89 (2C, C-2), 158.22 &162.13(C=Noxadiazole). HREIMs m/z = 357.1320 [M˙+] (calc. for C18H19N3O5, 357.1325).
General synthesis of 4-(((4-(5-((aryidene)amino)-1,3,4-oxadiazol-2-yl)benzyl)oxy) methyl)-2,6-dimethoxyphenol (7a-e)
To hot solution of 4-(((4-(5-amino-1,3,4-oxadiazol-2-yl)benzyl)oxy)methyl)-2,6-dimethoxy phenol (1 mmol) in glacial acetic acid-ethanol 1:1 (20 mL), 4-hydroxy substituted benzaldehyde (1 mmol) in ethanol (10 mL ) were added drop wise. The mixture refluxed for 18h. Then reduce the solvent to half under reduced pressure. After cooling the precipitate collected, washed with cold distilled water and recrystallized from suitable solvent.
Synthesis of 4-(((4-(5-((4-hydroxybenzylidene)amino)-1,3,4-oxadiazol-2-yl)benz yl)oxy)methyl)-2,6-dimethoxyphenol (7a)
Crude product recrystallized from aqueous ethanol to produce pale yellow precipitate, Yield 72%, Mp179-181°C, IR (KBr) vmax, 3548(OH phenol), 3087(CHAromatic), 1615 (CH=N), 1605 (C=NOxadiazole), 1600, 1985(C=C), 1250(C-N), 1203(C-O), 1099 (C-O-C); cm-1, 1H-NMR (DMSO-d6, δ): 3.65(s, 6H, 2× OCH3), 4.40 (s, 2H, OCH2), 4.49 (s, 2H, OCH2), 6.62 (s, 2H, H-3), 6.86( 2H, d, J 8.5, H-16), 7.86 (d, 4H, J 8.5,H-15), 7.91 (d, 2H, J 8.22,H-8), 8.2(d, 2H, J 8.2,H-9), 8.66(s, 1H, CH=N), 9.19(bs, 1H, OH), 9.96 (bs, 2H, OH), 13C-NMR (100 MHz, DMSO-d6): 56.12(2C, OCH3), 71.21(1C, CH2OCH2), 69.91(1C, CH2OCH2), 107.85(2C, C-3), 116.3(2C,C-16,C-18 ), 125.66(1C,C-10) 129.01(2C, C-9), 129.15(1C,C-14), 130.47(2C,C-15,C-19), 131.33(2C, C-8), 133.04(1C, C-4), 138.75(1C,C-1), 142.07 (1C,C-7) 151.89 (2C,C-2), 157(1C,C-17), 160.31(C=N), 160.93 &162.23(C=N oxadiazole). HREIMs m/z = 461.1581 [M˙+] (calc. for C25H23N3O6, 461.1587).
Synthesis of 4-(((4-(5-((4-hydroxy-3-methoxybenzylidene)amino)-1,3,4-oxadiazol-2-yl)benzyl)oxy)methyl)-2,6-dimethoxyphenol 7b
The crude mixture was purified by column chromatography using hexane- ethylacetate (5:1) as eluent to give a white amorphous solid. Yeild 69%, Mp 122-124°C. IR (KBr) vmax, 3608(OH phenol), 3083(CHAr), 1621 (CH=N), 1605 (C=Noxadiazole), 1594, 1983(C=C), 1231(C-N), 1193(C-O), 1109 (C-O-C); cm-1. 1H NMR (DMSO-d6, δ): 3.67(s, 6H, 2× OCH3), 3.88 (s, 3H, OCH3), 4.43 (s, 2H, OCH2), 4.55 (s, 2H, OCH2), 6.62 (s, 2H, H-3), 6.83 (d, 1H, J 7.78,H-18), 6.86( 2H,d, J 8.1,H-8), 7.23 (dd, 2H, J 8.24;1.8,H-19), 7.41 (s, 1H,H-15)) ,8.2( 2H, d, J 8.2,H-9), 8.59 (s, 1H, CH=N), 9.19(bs, 1H, OH), 9.96 (bs, 2H, OH). 13C-NMR (100 MHz, DMSO-d6): 56.11(2C, OCH3), 56.28(1C,OCH3) 71.21(1C, CH2OCH2), 69.91(1C, CH2OCH2), 107.85(2C, C-3), 110.32(1C,C-15), 115.85(1C,C-18), 124.22 (1C,C-10), 126.0(1C, C-19), 129.01(2C, C-9), 129.15(1C, C-14), 131.33(2C, C-8), 138.75(1C, C-1), 142.07 (1C, C-4), 148.5(1C,C-7), 151.89 (2C, C-2), 153.4(1C, C-16), 157(1C, C-17), 161.30(C=N), 162.53 &164.22(C=N oxadiazole). HREIMs m/z = 491.1690 [M˙+] (calc. for C26H25N3O7, 491.1693).
Synthesis of 4-(((4-(5-((4-hydroxy-3-ethoxybenzylidene)amino)-1,3,4-oxadiazol-2-yl)benzyl)oxy)methyl)-2,6-dimethoxyphenol 7c
Crude product recrystallized from chloroform-methanol to afford off white precipitate. Yield 65 %. Mp 148-150 °C. IR (KBr) vmax, 3595(OH phenol), 3077(CHAr), 2983, 2865 (CH aliphatic) 1618 (CH=N), 1607 (C=Noxadiazole), 1595, 1986(C=C), 1225(C-N), 1203(C-O), 1111 (C-O-C); cm-1. 1H-NMR (400MHz, DMSO-d6) ) 1.41 (t, 3H, J 6.9, O-CH2CH3), 3.71(s, 6H, 2× OCH3), 3.92 (q, 2H, J 7.0, O-CH2), 4.42 (s, 2H, OCH2), 4.45 (s, 2H, OCH2), 6.54 (s, 2H, H-3), 6.86 (d, 2H, J 8.2, H-18), 7.24 (dd, 1H, J 8.2; 1.7, H-19), 7.48(s, 1H, H-15), 7.71 ( 2H, d, J 8.24, H-8), 8.21( 2H, d, J 8.2,H-9), 8.57 (s, 1H, CH=N), 9.16(bs, 1H, OH), ), 9.78 (bs, 1H, OH),13C-NMR (100 MHz, DMSO-d6): 15.2 (1C, O-CH2CH3), 56.31(2C, OCH3), 64.2 (1C, OCH2), 71.21(1C, CH2OCH2), 69.91(1C, CH2OCH2), 107.85(2C, C-3), 110.19(1C, C-19), 116.25 (1C, C-15), 123.8 (1C, C-18), 126.44(1C, C-10), 129.01(2C, C-9), 129.15(1C, C-14), 131.33(2C, C-8), 133.04(1C,C-1), 138.75(1C,C-4), 142.07 (1C,C-7), 147.26 1C,C-16), 150.6 (1C,C-17) 151.89 (2C,C-2), 161.01(1C, C=N), 163.12 &164.03(C=N oxadiazole). HREIMs m/z = 505.1845 [M˙+] (calc. for C27H27N3O7, 505.1849).
Synthesis of 4-(((4-(5-((4-hydroxy-3,5-dimethoxybenzylidene)amino)-1,3,4-oxadiazol-2-yl)benzyl)oxy)methyl)-2,6-dimethoxyphenol 7d
Crude product recrystallized from ethylacetate-ethanol to afford pale yellow precipitate. Yield 62 %. Mp 166-168 °C. IR (KBr) vmax ,3610(OH phenol), 3082(CHAr), 2972, 2860 (CH aliphatic) 1614 (CH=N), 1605 (C=Noxadiazole), 1595, 1980(C=C), 1221(C-N), 1198(C-O), 1135 (C-O-C); cm-1, 1H-NMR (400MHz, DMSO-d6) δ:3.71(s, 6H, 2× OCH3), 3.85 (s, 6H, 2×OCH3) 4.42 (s, 2H, OCH2), 4.45 (s, 2H, OCH2), 6.57 (s, 2H, H-3), 7.19 (s, 2H, H-15,H-19), 7.75 (2H, d, J 8.23, H-8), 8.18 ( 2H, d, J 8.2, H-9), 8.68 (s, 1H, CH=N), 9.14 (2H, bs, 2×OH), 13C-NMR (100 MHz, DMSO-d6): 56.24(2C, OCH3), 56.36(2C, OCH3), 70.77(1C, CH2 OCH2), 68.92(1C, CH2OCH2), 105.86 (2C, C-3), 107.98(2C, C-15,C-19), 124.8(1C, C-10), 129.21(2C, C-9), 129.15(1C, C-14), 131.33(2C, C-8), 135.04(1C, C-4), 138.75(1C, C-7), 142.4 (2C, C-1, C-17) 151.89 (4C, C-2, C-16, C-18), 160.34(1C, C=N), 162.13&163,75 (C=Noxadiazole). HREIMs m/z = 521.1793 [M˙+] (calc. for C27H27N3O8, 521.1798).
Synthesis of 4-(((4-(5-((4-hydroxy-3,5-di-tert-butylbenzylidene)amino)-1,3,4-oxadiazol-2-yl)benzyl)oxy)methyl)-2,6-dimethoxyphenol 7e
The crude products recrystallized from Chloroform afforded pale yellow precipitate. Yield 59.7%, mp 204-206, IR (KBr) vmax, 3633.7 (OHphenol) 3058 (CHAr), 2960-2885 (CHalph), 1621(CH=N), 1601(C=Noxadiazole), 1597-1589 (C=C) and 1262(ArO-CH2) 1152(C-O-C). 1H NMR, (400 MHz, CDCl3,) δ: 1.42(s, 18H, 2×(t-Bu),3.71(s, 6H, 2× OCH3), 4.42 (s, 2H, OCH2), 4.45 (s, 2H, OCH2), 5.62 (bs, 1H, OH), 6.57 (s, 2H, H-3), 7.72 ( 2H, d, J 8.2, H-8), 7.91(s, 2H, H-15,H-19), 8.18( 2H, d, J 8.2, H-9), 8.59(s, 1H, CH=N), 9.14(bs, 1H, OH), 13C-NMR (100 MHz, DMSO-d6): 30.32(6C, 4(C(CH3)3), 34.56(2C, C(CH3)3), )) 56.12(2C, OCH3), 71.21(1C, CH2OCH2), 69.91(1C, CH2OCH2), 107.85(2C, C-3), 123.76 (2C, C-15, C-19) , 124.81 (1C, C10), 129.01(2C, C-9), 129.15(1C,C-14), 131.33(2C, C-8), 134.04(1C, C-4), 136.17(2C, C-16, C-18)138.44(1C,C-1) , 142.07 (1C, C-7) 151.89 (2C, C-2), 157.25 (1C, C-17), 161.44 (1C, C=N), 162.44&165.11 (C=N oxadiazole). HREIMs m/z = 573.2833 [M˙+] (calc. for C33H39N3O6, 573.2839).
Antioxidant
DPPH assay
The assay was performed as reported by Gerhauser et al. 27. Five microliters of the sample (dissolved in ethanol) was added into 195 μL of 100 μM DPPH reagent in ethanol (96%) and mixed in a 96-well plate. The intensity of the color was measured for 3 h at an interval of 20 min at 515 nm. Ascorbic acid and BHT were used as reference
FRAP assay
The FRAP assay was performed according to the Benzie and Strain 28method. The FRAP reagent was prepared by combining 300 mM acetate buffer and 10 mM 2,4,6-tripyridyl-s-triazine (TPTZ) solution in 40 mM HCl and 20 mM FeCl3·6H2O, in a ratio of 10:1:1. The FRAP reagent was incubated at 37 °C prior to use. Ten micro liters of the sample was reconstituted in the carrier (solvent or ultra pure water) and mixed with 300 μL of FRAP reagent. The mixture was incubated at 37 °C for 4 min in a micro plate reader. The absorbance of the complex was 593 nm. The FRAP value can be calculated using the following equation29:
FRAP = [(0–4 min ∆A 593 nm of test sample)/(0–4 min ∆A 593 nm of standard)]
× [standard] (µM) × Y × 1000
Where Y is the absorbance of the spectrophotometer.
Results and Discussion
Chemistry
The new compounds (7a-7e) were synthesized from multi-stapes. Scheme 1 displayed the synthesise of 5-dimethyl-4-((trimethylsilyl)oxy)benzyl alcohol (2) which is used as starting material to synthesis of 4-(((3,5-dimethoxy-4-((trimethylsilyl)oxy)benzyl)oxy)methyl) benzonitrile (3) as depicted in Scheme 2.
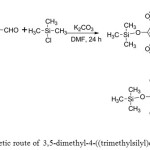 |
Scheme1: Synthetic route of 3,5-dimethyl-4-((trimethylsilyl)oxy)benzyl alcohol.
|
Compound 3 was converted to the corresponding acid 4 then the acid to the corresponding hydrazide (5). The hydrazide cyclized to the 5-amino-1,3,4-oxadiazole 6. The 5-amino-1,3,4-oxadiazole were reacted with several substituted of 4-hydroxybenzylaldehyde (Table 1 ) as shown in Scheme 2.
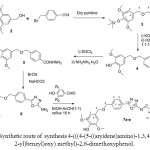 |
Scheme2: Synthetic route of synthesis 4-(((4-(5-((aryidene)amino)-1,3,4-oxadiazol-2-yl)benzyl)oxy) methyl)-2,6-dimethoxyphenol. Click here to View scheme |
Table 1. The substituent group and selected properties of synthesized compound 7a-7e
|
No. |
R1 |
R2 |
Yield % |
M.P. ̊ C |
HREIMS calc. |
HREIMS Found |
|
7a |
H |
H |
72 |
179-181 |
461.1587 |
461.1581 |
|
7b |
H |
OCH3 |
69 |
122-124 |
491.1693 |
491.1690 |
|
7c |
H |
OEt |
65 |
148-150 |
505.1849 |
505.1845 |
|
7d |
OCH3 |
OCH3 |
62 |
166-168 |
521.1798 |
521.1793 |
|
7e |
C(CH3)3 |
C(CH3)3 |
60 |
204-206 |
573.2839 |
573.283 |
All synthesized compounds were characterized from their IR, 1H-NMR, 13C-NMR spectrum beside the EIMS and HRMIS.
The IR spectrum of compound 3, showed an interesting peak at 2248 cm-1 for C≡N which is disappearing from compound 4 and a new peak of C=O was appeared at 1673 cm-1, as well interesting peak of C=N of the oxadiazole ring for compound 6 was appearing at 1609 cm-1. Moreover, the C=N of Schiff bases of compounds 7a-7e were appeared at 1614-1621 cm-1 beside the C=N of the oxadiazole ring. The 1H-NMR spectrum of compound 2 showed the disappearing of the proton of aldehyde from 9.85 ppm and appearing of two protons of CH2 beside the OH which represents the successful reduction of aldehyde group. Furthermore, the new two doublets for compound 3 at 7.41 and 7.49 ppm ( H-8, H-9 )with copling constant (J) 8.2 and 7.9 Hz respectively represented successfully of reaction compound 2 with 4-(bromo methyl)benzonitrile. As well 1H-NMR of compound 4 showed disappearing of trimethyl lsilyl grop from 0.18 ppm and appears of the broad singlet of OH for the converting the nitrile group to carboxylic acid. The 1H-NMR spectrum of compound 5 showed the disappearing of H of carboxylic and appeared of new two board singlet of NH2 and NH at 4.74 and 8.83 ppm respectively, while compound 6 showed disappear these peaks and appeared new signal at 7.51 which is represented the NH2 attached the oxadiazole ring. The 1H-NMR of compounds 7a-7e showed appearance of new peaks represented the Schiff base (Imin proton) at 8.57-8.68 ppm. The peaks of the substituted 4-hydroxylphenol appeared in their expected areas (see the experimental section). The 13C-NMR spectrum of compound 2 showed the carbons of trimethylsilyl at -0.05 as well the carbon of CH2-OH. While the 13C-NMR spectrum of compound 3 displayed two carbons of CH2OCH2 at 70.76 & 71.14 beside the C≡N at 101.22 also the carbons of benzonitrile ring. Disappearing of carbon C≡N and raised a new carbon of C=O that Enhances the evidence of successful convert compound 3 to 4 besides the IR and 1H-NMR. The 13C-NMR of compound 6 showed two carbons of C=N of oxadiazole ring instead of the carbon of the carbonyl group. The 13C spectra of compounds 7a-7e were showed an interested peak of CH=N beside the carbons of the substitute 4-hydroxyphenol. As well as all the substituent group for 7b-7e were appeared in the expected range. The EIMS spectra showed the molecular ion M•+ for all compounds and the base peak (100%) as well the HREIMs value was confirmed the accurate mass and the molecular formula as depicted in Table 1.
Antioxidant activity
The synthesized compounds (7a-7e) were exhibited high antioxidant ability in both assaysBased on the type of substitute group of phenol. Compound 7e, which I possess 2,6 di-tert-butylphenol group shows antioxidant ability slightly higher than ascorbic acid in both assays (DPPH and FRAP) as shown in Figure 1
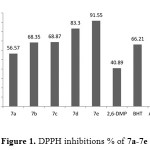 |
Figure1: DPPH inhibitions % of 7a-7e Click here to View figure |
While, compound 7d which is posses 2,6-di methoxy phenol group exhibited antioxidant ability higher than BHT also, all compounds showed antioxidant activity more than 2,6-dimethoxy phenol it self. The antioxidant of the substituted phenol followed the following sequence: 2,6-di-tert-butyl>2,6-dimethoxy>2-methoxy≈2-ethoxy> non substituted phenol. These results are in agreement with many articles xxx that the more hindered phenol increase the antioxidant stability. Further more, we assume that the long chain resonance between the substituted phenol and the main group (CH=N) attached the oxadiazole ring after donating a proton could enhance the free radical stability, which can enhance the free-radical scavenging ability. As shown in Figure 2.
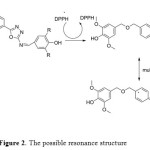 |
Figure2: The possible resonance structure Click here to View figure |
The FRAP value was higher than DPPH when comparing the results with the references and this difference could be attributed to the different mechanisms for FRAP and DPPH. FRAP involves a single electron transfer mechanism, whereas DPPH assay depends on the H-atom transfer mechanism30 beside the hindrances around the phenol. Figure 3 displayed the FRAP value of the synthesized compound 7a-7e
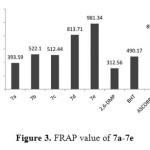 |
Figure3: FRAP value of 7a-7e Click here to View figure |
Conclusions
A series of new Schiff base compounds from 5-amino-1,3,4-oxadizole incorporating hindered phenol moieties were successfully synthesized and characterized. All of the new compounds were screened for antioxidant activity using the FRAP and DPPH assays. The antioxidant ability of these compounds increases with increasing the hindered phenol.
Conflicts of Interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper
Acknowledgements
The author would like to thank the University of Malaya for running the NMR We are also grateful to Mr. Mohammed Farouq Halabi, Department of Biomedical Science, Faculty of Medicine, University of Malaya for his great help with screening the antioxidant and Dr.Raied M. Shakir for his contributions as well, we would like to Acknowledge NUS, Singapore for running the EIMS and HREIMS. Also, we would like to thank the University of Baghdad for supporting this study and provide the grant for this study.
References
- Shacter, E. Drug Metabolism Reviews 2000, 32, 307.
- Chatterjee, R.; Bandyopadhyay, U.; Mazumdar, A.; Banerjee, R. K. Biochem Pharmacol. 1996, 52, 1169.
- Sangeetha, P.; Das, U. N.; Koratkar, R.; Suryaprabha, P. Free Radic Biol Med. 1990, 8, 15.
- Kar, S., Subbaram, S., Carrico, P M., Melendez, J A. Respir Physiol Neurobiol. 2010, 174, 299.
- Khalil, Z.; Khodr, B. Free Radic Biol Med. 2001, 31, 430.
- Mullican, M. D.; Wilson, M. W.; Connor, D. T.; Kostlan, C. R.; Schrier, D. J.; Dyer, R. D. J. Med. Chem 1993, 36, 1090.
- Ye, X.; Zhou, W.; Li, Y.; Sun, Y.; Zhang, Y.; Ji, H.; Lai, Y. Cancer Chemotherapy and Pharmacology 2010, 66, 277.
- YEN, C.-Y. H. A. G.-C. J. Agric. Food Chem. 2002, 50, 2993.
- Shakir, R. M.; Ariffin, A.; Abdulla, M. molecules 2014, 19, 3436.
- Jeong , J.-M.; Kang, S.-K.; Lee, I.-H.; Lee, J.-Y.; Jung, H.; Choi, C.-H. J Pharm Pharmaceut Sci 2007, 10, 537.
- Edge, R.; McGarvey, D. J.; Truscott, T. G. J Photoch Photobio B 1997, 41, 189.
- Sreeramulu, J.; Ashokgajapathiraju, P. Orient J Chem 2014, 30, 651.
- SHAH, H. P., SHAH, B. R., BHATT, J. J., DESAI, N. C., TRIVEDI, P. B. and UNDAVIA, N. K. Indian J. Chem 1998, 37, 180.
- de Oliveira, C. S.; Lira, B. F.; Barbosa-Filho, J. M.; Lorenzo, J. G. F.; de Athayde-Filho, P. F. molecules 2012, 17, 10192.
- Musad, E. A.; Mohamed, R.; Ali Saeed, B.; Vishwanath, B. S.; Lokanatha Rai, K. M. Bioorganic & Medicinal Chemistry Letters 2011, 21, 3536.
- Radhika Maheshwari, P. C., Shubhini Saraf Medicinal Chemistry Research 2010, 1.
- Ogiri, S.; Ikeda, M.; Kanazawa, A.; Shiono, T.; Ikeda, T. Polymer 1999, 40, 2145.
- Liu, H.; Fu, Z.-e.; Xu, K.; Cai, H.-l.; Liu, X.; Chen, M.-C. Materials Chemistry and Physics 2012, 132, 950.
- Shan, X.; Ibrahim, A. O.; Zhou, Y.; Zhang, H.; Ma, J.; Jiang, F.; Hong, M. Inorganic Chemistry Communications 2012, 22, 149.
- Kim, S.-H.; Gwon, S.-Y.; Burkinshaw, S. M.; Son, Y.-A. Dyes and Pigments 2010, 87, 268.
- Chandra, M.; Sahay, A. N.; Pandey, D. S.; Tripathi, R. P.; Saxena, J. K.; Reddy, V. J. M.; Carmen Puerta, M.; Valerga, P. Journal of Organometallic Chemistry 2004, 689, 2256.
- Bodtke, A.; Pfeiffer, W.-D.; Ahrens, N.; Langer, P. Bioorganic & Medicinal Chemistry Letters 2004, 14, 1509.
- Kaya, İ.; Cihangiroğlu, N. Journal of Polymer Research 2004, 11, 37.
- Khodair, A. I.; Bertrand, P. Tetrahedron 1998, 54, 4859.
- Huihui Ti; Qing Li; Ruifen Zhang; Mingwei Zhang; Yuanyuan Deng; Zhencheng Wei; Jianwei Chi; Yan Zhang Food Chemistry 2014, 159 166.
- Fausta Natella, M. N., Maurizio Di Felice, and Cristina Scaccini*; Free Radical Research Group, N. I. o. N., Roma, Italy J. Agric. Food Chem. 1999, 47, 1453.
- Gerhäuser, C.; Klimo, K.; Heiss, E.; Neumann, I.; Gamal-Eldeen, A.; Knauft, J.; Liu, G.-Y.; Sitthimonchai, S.; Frank, N. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 2003, 523-524, 163.
- Benzie, I. F.; Strain, J. J. Anal. Biochem. 1996, 239, 70.
- Rustaiyan, A., Javidnia, K., Farjam, M H., Aboee-Mehrizi, F., E. J Med Plants Res. 2011, 5, 4251.
- Huang, D.; Ou, B.; Prior, R. L. Journal of Agricultural and Food Chemistry 2005, 53, 1841.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


