Synthesis of New Organoselenium Compounds Containing Nucleosides as Antioxidant
Laila M. Break1, Mahmoud A. Mohamed1,2 and Shams H. Abdel-Hafez1,3
1Department of Chemistry, Faculty of Science, Taif University, Taif, Saudi Arabia
2Biochemistry department, Faculty of Agriculture, Cairo University, Cairo, Egypt
3Department of Chemistry, Faculty of Science, Assiut University, Assiut, Egypt
DOI : http://dx.doi.org/10.13005/ojc/300423
Article Received on :
Article Accepted on :
Article Published : 18 Nov 2014
Selenium containing nucleosides derived from some heterocyclic moieties such as Pyridineselenol, and pyridazineselenol is described herein. Ribosylation of selenol compounds were prepared in good yield by silyation of selenol derivatives with 1-O-acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose followed by debenzoylation to afford the corresponding free N-nucleosides b and a-1-(2,3,5-trihydroxy-
KEYWORDS:1-O-Acetyl-2,3,5-trihydroxy-β-D-ribofuranose; Nucleosides; Selenium ; Pyridineselenol; pyridazineselenol; antioxidants
Download this article as:| Copy the following to cite this article: Break L. M, Mohamed M. A, Abdel-Hafez S. H. Synthesis of New Organoselenium Compounds Containing Nucleosides as Antioxidant. Orient J Chem 2014;30(4). |
| Copy the following to cite this URL: Break L. M, Mohamed M. A, Abdel-Hafez S. H. Synthesis of New Organoselenium Compounds Containing Nucleosides as Antioxidant. Available from: http://www.orientjchem.org/?p=5436 |
Introduction
From the literature survey indicates that few publications have mentioned the incorporation of selenium atom into nucleosides [1,2]. In this paper [1] describe the synthesis of selenium- and tellurium-containing nucleosides, derived from uridine which was prepared in a concise and short synthetic route in good yields, by nucleophilic substitution of a tosylate group by organoselenium nucleophiles. On the other hand in previous work in our laboratory [2] describes the synthesis of selenium-containing nucleoside analogues, derived from some heterocyclic moieties such as pyridineselenol, pyridazineselenol and quinolineselenol derivatives which indicated that one of pyridine-moiety 2-(1,3- dihydroxypropylselenyl)-4,6-dimethylpyridine-3- carbonitrile which bearing two hydroxyl groups, and three of pyridazine-moieties 3-(3-hydroxypropyl selenyl)-5,6-diphenylpyridazine-4-carbonitrile, 3-(1,3-dihydroxy propyl selenyl)-5,6-diphenylpyridazine-4-carbonitrile and 3-(oxiran-2-ylmethyl selenyl)-5,6-diphenyl- pyridazine-4-carbonitrile respectively have definite antioxidant effect.
Stimulated by our recent work on the synthesis of selenium containing nucleoside analogues [2], sulfa drugs, [3] and the synthesis of selenium containing amino acid analogues, [4] we decided to expand our interest to the introduction of an organoselenium compounds in the nucleoside framework and screened their biological activity as antioxidants.
Results and Discussion
Chemistry
Ribosylation of 1a, b were achieved by refluxing in hexamethyldisilazane (HMDS) to give the silylated derivatives 2a, b. The latter was stirred with 1-O-acetyl-2,3,5-O-benzoyl-β-D-ribofuranose (3) in the presence of dry 1,2-dichloroethane as a solvent using trimethylsilyl trifluoromethane sulfonate (TMS Triflate) (CF3SO2OSiMe3) as a catalyst as according to the method of Vorbruggen et al [5] to give the corresponding mixture of β-anomeric protected N-nucleoside derivatives and a-anomeric protected N-nucleoside. Separation of the mixture of β and a anomers (4a, 5a) and (4b, 5b) was carried out by column chromatography in yields ranging from 25-75 % of the corresponding benzoylated nucleosides (4a, 5a, 4b, and 5b) respectively (Scheme 1).
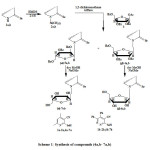 |
Scheme1: Synthesis of compounds (4a,b- 7a,b) Click here to View scheme |
Debenzoylation of compounds (4a, 5a, 4b and 5b) were performed by using methanolic sodium methoxide solution following Zemplen et al. method [6] to afford the free nucleosides (6 a, 7a, 6b and 7b) respectively.
The chemical structures of the nucleoside derivatives 4a, 4b, 5a, 5b, 6a, 6b, 7a and 7b were established and confirmed on the basis of their elemental analyses and spectral data (IR, 1H and 13C NMR) (see the Experimental section).
The IR spectra of compounds (4a, 5a, 4b and 5b) were observed at v 2210 cm−1 due to CN group and stretching vibration frequencies of the benzoyl carbonyl groups C=O appeared at v 1740, 1730, 1727 and 1724 cm-1 of compounds 4a, 5a, 4b and 5b respectively. In addition signals at v=1625, 1620 cm−1 for the C=N group of compounds 4a, 5a, and at v 1630 cm-1 of compounds 4b and 5b.
The IR spectra and the most important peaks for compounds (6a, 7a, 6b and 7b) were observed at v 3400-3450 cm−1 due to (OH group) for compounds 6a and 6b respectively and signals at v 3380 cm−1 due to (OH group) for compounds 7a and 7b.
The 1H NMR spectra of compounds (4a, 5a, 4b and 5b) showed a doublet ranging from δ 5.55 – 6.39 ppm , with spin–spin coupling constant (∫1′,2′) equal to 7.5 Hz for proton (H-1′) which confirms the β-anomeric configuration of compound 4a and 4b respectively, while the data showed that a doublet signals at δ ranging from 5.80 – 6.39 ppm , with spin–spin coupling constant (∫1′,2′) ranging from 4.5-5 Hz for proton (H-1′) assigned to the a -anomeric configuration of compound 5a and 5b respectively.
The 1H NMR spectra of compounds (6a, 7a, 6b and 7b) showed a doublet ranging From δ 6.20-6.08 ppm with (∫1‘,2‘) equal to 7-7.5 Hz for for proton (H-1′) which confirms the β-anomeric configuration of compound 6a and 6b. While signals at δ ranging from 6.08 – 6.22 ppm , with spin–spin coupling constant (∫1‘,2‘) ranging from 5.5-5 Hz for proton (H-1′) assigned to the a -anomeric configuration of compound 7a and 7b respectively.
The 13C NMR spectrum of compounds (4a, b, 5a, b, 6a, b and 7a, b) showed the most signals at δ C 13.4 and 18.6 for two groups of CH3 for compounds 4a and 5a. The five signals at δ C 93.2, 84.2, 78.1, 71.4 and 59.2 were assigned to C-1′, C-2′ , C-3′ , C-4′, and C-5′ of the sugar moiety, respectively. Data showed that at δ C 118.2 due to (CN), 120.0–135.0 (Ar), 151.2, 160.2, 168.0 (C=O); 175.0 (C=Se) of compound 4a and 175.9 (C=Se) of compound 4b. The 13C NMR spectrum of compounds 7a and 7b showed signals: 22.63, 29.70, 52.15, 63.42, 76.12, 79.11, 84.71, 117.786, 128.39, 129.74, 132.09, 133.26, 169.79 for 7a and signals at δ 52.12, 64.50, 76.12, 79.11, 89.51, 117.53, 127.34, 128.28, 128.36, 128.65, 128.88, 128.99, 129.57, 132.06, 132.92, 133.23, 134.13, 167.72, 169.27 for 7b.
Finally the Mass spectra and Elemental analysis are in agreement and confirmed of all new compounds.
Biological activity
Toxicity studies
Toxicity parameters including LD50; GPT and LDH activities were determined and ranged in normal limits compared to infected untreated group with concentrations range up to 1000 mg kg- b.wt. The GPT, an enzyme which allows determining the liver function as indicator on liver cells damage and LDH enzyme is often used as a marker of tissue breakdown (Butt et al., 2002) [6].
Antioxidant activity evaluation.
Hepatic GSH-Rd and serum activities of SOD, GSH-S-transferase levels were measured as an indicator of antioxidant activity and result are present in Table 1. SOD and GSH-S-transferase are antioxidant enzymes that protect cells from oxidative stress of highly reactive free radicals and induces on the generation of free radicals in living cells. Result indicated that significant increasing (p < 0.05) in SOD, and GST activities in the treated groups at doses of 100 and 200 mg/kg compared to un-treated control group. No significant deference found between used doses (100 and 200 mg/kg) with all tested compounds. The highest SOD and GST, activities rather than GSH-Rd levels was monitored in animals treated with compounds 6a and 7b. (See Table 1, Fig. 1).
Table 1. Effects of synthesized compounds (1a; 6a; 7a; 1b; 6b and 7b) on SOD; GST activities and GSH-Rd levels.
| Design of treatment | SOD(Units/mg protein) | GST (µmol /mgprotein) | GSH-Rd (mg/g protein) |
| Untreated group,Group 1 | 35.13 ± 1.25a,b | 5.13 ± 0.11a,b | 4.21 ± 0.21a,b |
| Compound 1a (100 mg/kg) (Group 2) | 35.87 ± 0.51 | 5.84 ± 0.11 | 4.10 ± 0.10 |
| Compound 1a (200 mg/kg) (Group 3) | 32.21 ± 1.93 | 6.13 ± 0.25 | 5.01 ± 0.32 |
| Compound 1b (100 mg/kg) (Group 4) | 34.39 ± 1.13 | 5.10 ± 0.61 | 4.91 ± 0.27 |
| Compound1b (200 mg/kg) (Group 5) | 33.90 ± 1.21 | 5.82 ± 0.18 | 5.11 ± 0.22 |
| Compound 6a (100 mg/kg) (Group 6) | 39.01 ± 1.15a,b, | 3.91 ± 0.26a,b | 5.85 ± 0.13a,b |
| Compound 6a (200 mg/kg) (Group 7) | 38.90 ± 1. 01 a,b, | 6.19 ± 0.10a,b | 5.39 ± 0.11a,b |
| Compound 6b (100 mg/kg) (Group 8) | 32.49 ± 2.10 | 5.01 ± 0.72b | 4.01 ± 0.12 |
| Compound 6b (200 mg/kg) (Group 9) | 33.54 ± 1.42 | 4.45 ± 0.11 | 3.92 ± 0.15 |
| Compound 7a (100 mg/kg) (Group 10) | 34.54 ± 2.10 | 5.12 ± 0.32 | 4.78 ± 0.17 |
| Compound 7a (200 mg/kg) (Group 11) | 31.97 ± 1.79 | 5.16 ± 0.40 | 4.98 ± 0.13 |
| Compound 7b (100 mg/kg) (Group 12) | 41.54 ± 1.15a,b | 7.12 ± 0.81a,b | 6.78 ± 0.14a,b |
| Compound 7b (200 mg/kg) (Group1 3) | 39.17 ± 1.02a,b | 6.82 ± 0.42a,b | 6.18 ± 0.08a,b |
| Vitamin E (100 mg/ kg) (Group 14) | 45.17 ± 1.13 | 8.12 ± 0.11 | 7.37 ± 0.31 |
Values are mean ± SD, n = 6, a b the difference is significant (p>0.05) in a column between treated and untreated control group 1.
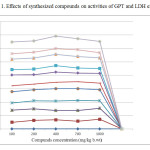 |
Figure1: Effects of synthesized compounds on activities of GPT and LDH enzymes. Click here to View figure |
Experimental
General
Melting points were determined by using the Kofler melting point apparatus, and were uncorrected. IR (KBr, cm−1) spectra were recorded on a Pye-Unicam SP3–100 instrument at Taif University. 1H NMR spectra were obtained on a Varian (400 MHz) EM 390 USA instrument at King Abdel-Aziz University by using TMS as internal reference.13 C NMR spectra were recorded on a JNM-LA spectrometer (100 MHz) at King Abdel-Aziz University, Saudi Arabia. Elemental analyses were obtained on an Elementar Vario EL 1150C analyzer. Mass spectra were recorded on a JEOL-JMS-AX 500 at Cairo National Research Center, Cairo, Egypt. Purity of the compounds was checked by thin layer chromatography (TLC) using silica gel plates.
Synthesis of a and b-1-(-(2,3,5-tri-O-benzoyl-β-D-ribofuranosyl)seleno derivatives (4a,b , 5a,b)
3.2.1. Ribosylation of 2-seleno-4,6-dimethylpyridine-3-carbonitrile and 3-seleno-5,6-diphenylpyridazine-4-carbonitrile (1a,b). Synthesis of b and a-1-(2,3,5-tri-O-benzoyl-β-D-ribofuranosyl)-2-seleno-4,6-dimethylpyridine-3-carbonitrile (4a, 5a); β and a-1-(2,3,5-tri-O-benzoyl-β-D-ribofuranosyl)-3-seleno-5,6-diphenylpyridazine-4-carbonitrile (4b, 5b).
General Procedure
A mixture of 2-seleno-4,6- dimethylpyridine-3-carbonitrile or 3-seleno-5,6-diphenylpyridazine-4-carbonitrile (1a,b) (0.02 mol) and hexamethyl di- silazane (20 ml) was heated under reflux for 24h with a catalytic amount of ammonium sulfate (0.01g). After that, the clear solution was cooled and evaporated till dryness to give the silyated derivative (2a,b), which directly was dissolved in 20 ml of dry 1,2-dichloroethane and then 1-O-acetyl-2,3,5-tri-O-benzoyl-β -D-ribofuranose (3) (5.05 g, 0.01 mol) was added. The mixture was added dropwise onto a mixture of (10 ml trimethylsilyl trifluoromethanesulfonate (Triflate) in dry 1,2-dichloroethane (50 ml)). All mixture was stirred at room temperature for 24 h, and then washed with a saturated solution of aqueous sodium bicarbonate (3 × 50 ml), washed with water (3 × 50 ml), and dried over anhydrous sodium sulfate. The solvent was removed in vacuum and the residue was chromatographic on silica gel with chloroform: ethyl acetate (9: 1) as eluent to afford a white crystal from pure anomeric b and colorless crystals from a anomeric (4a,b and 5a,b) respectively.
Compound (pydBzb) (4a)
Yield (42%), m.p. 160-161∘C; IR (KBr) vcm−1: 2217 (CN), 1730 (CO). 1H NMR (CDCl3): v 3.19 (s, 3H, CH3); 2.20 (s, 3H, CH3); 3.98-4.10 (m, 2H, 2H-5′); 4.90 (m, 1H, 1H-4′), 5.25–5.30 (m, 1H, H-3′), 5.38–5.40 (m, 1H, H-2′), 5.55 (d, 1H, H-1′, v1‘,2‘ = 7.5Hz), 6.32 (s, 1H, CH-pyridine), 7.33–8.05 (m, 15H, aromatic protons); 13CNMR (CDCl3): 16.2, 20.3, 59.2, 71.4, 78.1, 84.2, 93.2, 118.2, 120.0–135.0, 151.2,160.2, 168.0; 175.0 (C=Se). Anal. Calcd. for C34H28N2O7Se (655.56): C, 62.29; H, 4.31; N, 4.27 (%); Found: C, 62.00; H, 4.11; N, 4.14 (%).
Compound (pydBza) (5a)
Yield (28%), m.p. 203-204∘C; IR (KBr) v cm−1: 2210 (CN), 1730 (CO). 1H NMR (CDCl3): δ 3.12 (s, 3H, CH3); 2.23 (s, 3H, CH3); 3.46-3.48 (m, 2H, 2H-5′), 4.60–4.95 (m, 1H, H-4′), 5.15-5.30 (m, 1H, H-3′), 5.40–5.60 (m, 1H, H-2′), 5.80-6.94 (d, 1H, H-1′, ∫1‘,2‘ = 4.5Hz), 6.30 (s, 1H, CH-pyridine), 8.00–7.45 (m, 15H, Ar-H); 13C NMR (CDCl3): 13.4, 18.6, 59.5, 71.2, 78.1, 84.0, 93.2, 121.4, 125.0–140.2, 151.2, 159.2, 168.0, 175.2 (C=Se); Anal. Calcd. for C34H28N2O7Se (655.56): C, 62.29; H, 4.31; N, 4.27 (%); Found: C, 62.15; H, 4.22; N, 4.10 (%).
Compound (PydzBzb) (4b)
Yield (47%), m.p. 185–187∘C; IR (KBr) v cm−1: 2210 (CN), 1730(CO), 1630 (C=N); 1H NMR (CDCl3): δ4.23 (d, 1H, H-4′ ∫1‘,2‘ = 7.5Hz); 4.58-4.57 (m, 2H,H-5′), 4.64 (d, 1H,H-3′, ∫1′,2’= 7.5Hz); 5.65 (d, 1H, H-2′, ∫1‘,2‘= 7.5Hz), 6.39 (d, 1H, H-1′, ∫1‘,2‘= 7.5Hz), 7.53-7.00-7.45 (d, 10H,aromatic protons, ∫ = 8.0Hz), 8.05–7.81 (m, 15H, Ar-H); 13CNMR (CDCl3): 59.1, 71.5, 78.3, 84.2, 93.0, 121.2, 125.0–140.0,151.2, 162.1, 168.4, 175.9 (C=Se); Anal. Calcd. for C43H31N3O7Se (780.68): C, 66.15; H, 4.00; N, 5.38 (%); Found: C, 66.08; H, 3.99; N, 5.10 (%).
Compound (PydzBza) (5b)
Yield (30%), m.p. 168-169∘C; IR (KBr)
Conclusion
Silyation of selenol derivatives with 1-O-acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose followed by debenzoylation affording the corresponding free N-nucleosides compounds b and a-1-(2,3,5-trihydroxy-b-D-ribofuranosyl)-2-seleno-4,6-dimethylpyridine-3-carbonitrile (6a,7a); b and a-1-(2,3,5-trihydroxy-b -D-ribofura- nosyl)-3-seleno-5,6-diphenylpyridazine-4-carbonitrile (6b,7b) respectively. Compounds 6a and 7b showed to be more active as antioxidant with a better performance of scavenging ability than other compounds. The SOD activity of these molecules was compared with standard antioxidant (vitamin E). Selenonucleoside compounds are active sites of a large number of selenium dependent enzymes, such as antioxitant enzymes [Spallholz J.E 1994] [7]. The configuration structure of compounds
6a and 7b may more suitable for SOD and GST enzymes active center, so these compounds induce the antioxidants enzymes activity.
References
- L. Antonio Braga, a. Wolmar Severo Filho, S. Icardo Schwab, E. D. Oscar Rodrigues, D. Lucian, C. Hugo Braga, S. Diogo Ludtke, Tetrahedron lett. 2009, 50, 3005-3007
- Sh. H. Abdel-Hafez, H. A. Saad, M. A. Mohamed. Der Pharma Chemica, 2012, 4(3):915-925
- Sh. H. Abdel-Hafez. Rus. J. Bioorg. Chem., 2010; 36(3): pp. 370–376
- Sh. H. Abdel-Hafez, Saad, H.A., Aly, M.R.E. Rus. J. Bioorg. Chem., 2011, 37(3), pp. 261-269.
- H. Vorbruggen, K. Krolikiewicz, and B. Bennua, Chem. Ber.,1981, 114, 1234
- A.A. Butt, S. Michaels, D. Greer, R. Clark, P. Kissinger and D.H. Martin, AIDS Read, 2002, 12: 317-321
- J.E. Spallholz .Free Radical Biol. Med., 1994, 17, pp. 45–64
- Ghosh, M.N., 2nd Edn., Scientific Book Agency, Kolkatta, ISBN: 81-902965-0-7, 1984, pp: 175-176.
- S. Reitman, and Frankel, S., Am. J. Clin. Pathol., 1957, 28: 56-56.
- H. U. Bergmeyer (Ed.). Verl. Chemie, Weinheim., 1974 V.76, Issue 5, P. 472
- S. Rai, W. Atul , M. Kakal, P. Bishn. and M.Pulok., J. of Ethnopharma. 2006, 104, 322-327.
- W.H. Habig, M.J. Pabst, W.B. Jakoby, J. Biol. Chem.; 1974, 249(22–25): 7130–7139.
- Kakkar, P., Das, B., Visvanathan, P.N., Indus. J. Biochem. and Biophys., 1972, 197, 588–590.
- G.L.Ellaman, Arch. Biochem. and Biophys. 1959. 82, 70-72
- M.M. Bradford, Anal.Biochem., 1976, 72: 248 – 254

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


