Theoretical Study Intramolecular Hydrogen Bond In Acetylacetone 3-Substituted Derivatives: NMR, NBO Analysis And Thermo-Chemical Investigation
Seyed Abdollah Seyed Katouli1*, Masoome Sheikhi2, Davood Sheikh3, Sayyed Faramarz Tayyari 4
1Department of Chemistry, Aliabad Katoul Branch, Islamic Azad University, Aliabad Katoul, Iran. 2Department of Chemistry, Gonbad Kavoos Branch, Islamic Azad University, Gonbad Kavoos, Iran 3Young Researchers & Elites Club, Hamedan Branch, Islamic Azad University , Hamedan, Iran. 4Department of Chemistry, University of Ferdowsi, Mashhad 91774-1436, Iran.
DOI : http://dx.doi.org/10.13005/ojc/290338
Article Received on :
Article Accepted on :
Article Published : 28 Oct 2013
The strength of the O–H···O hydrogen bridge in acetylacetone (AA) and acetylacetone derivatives is depending on the nature and size of the substitute groups and on the substitution position. We investigated the effect of electron-pull and electron-donor substitute on the formation of intramolecular hydrogen bonding at 3-substituted acetylacetone derivatives such as nitroacetylacetone (NAA) and methylacetylacetone (MAA). In this research NAA and MAA structures were fully optimized with B3LYP/6-31G*, 6-31G** and 6-311G**. From the electronic data we found that intramolecular hydrogen bonding in NAA is stronger than MAA.
KEYWORDS:intramolecular hydrogen bond;acetylacetone;electronic data parameters
Download this article as:| Copy the following to cite this article: Katouli S. A. S, Sheikhi M, Sheikh D, Tayyari S. F. Theoretical Study Intramolecular Hydrogen Bond In Acetylacetone 3-Substituted Derivatives: NMR, NBO Analysis And Thermo-Chemical Investigation. Orient J Chem 2013;29(3). doi : http://dx.doi.org/10.13005/ojc/290338 |
| Copy the following to cite this URL: Katouli S. A. S, Sheikhi M, Sheikh D, Tayyari S. F. Theoretical Study Intramolecular Hydrogen Bond In Acetylacetone 3-Substituted Derivatives: NMR, NBO Analysis And Thermo-Chemical Investigation. Orient J Chem 2013;29(3). Available from: http://www.orientjchem.org/?p=337 |
Introduction
Hydrogen bonding is one of the most important phenomena in chemistry because it is crucial to understand many different interactions both in the gas phase and in condensed media (1,2). In a particular arrangement, which is represented by the intramolecular hydrogen bonds, two ends of the same molecule interact, resulting in a ring like structure. The properties of intramolecular hydrogen bond very often differ from those of intermolecular hydrogen bond and a number of regularities and relationships resulting from a general theory of hydrogen bond cannot be fulfilled in the case of intramolecular hydrogen bond. Intramolecular hydrogen bonding has a large impact on the reactivity of molecules.
One of the more significant structures capable of bearing hydrogen bonds is the O–H···O unit, which is the most widely studied and documented in this respect (3–6). Acetylacetone (pentane-2,4-dione, here labelled AA) is one of the simplest members of β-diketones, which has been extensively studied both experimentally and theoretically (7-10). AA is postulated to have unusually strong H-bonds (O–H···O type) in their cyclic, conjugated enolic forms (Figure 1). The strength of the O–H···O hydrogen bridge in acetylacetone and acetylacetone derivatives is depending on the nature and size of the substitute groups and on the substitution position (5,6,11). Several experimental data suggest that the strength of such a bridge is enhanced when the H atom in position 3 is substituted by electron-withdrawing groups (12), and it increases strongly when very cumbersome substituents are involved because steric interactions push the two oxygen atoms closer to each other (13–17).
The ab initio calculations using the Møller–Plesset approach indicate that strengthening of the hydrogen bridge, on passing from the parent (AA) to the 3-substituted derivatives, is not so relevant as expected on the ground of literature data, the maximum increase being about 21 kJ mol−1 (in 3-t-butyl-acetylacetone) (18).
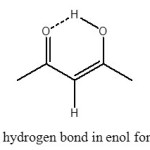 |
Figure 1. intramolecular hydrogen bond in enol form of Acetylacetone (AA) Click here to View figure |
Ab initio calculations were carried out for nitroacetylacetone (NAA) and methylacetylacetone (MAA) using the Gaussian 98 program (Figure 2).
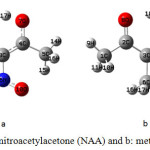 |
Figure 2. Structure of a: nitroacetylacetone (NAA) and b: methylacetylacetone (MAA). Click here to View figure |
We optimized the geometries of NAA and MAA to calculate ground state properties Becke’s three parameter hybrid method (Becke, 1988) with the Lee et al. (1988) (19) correlation functional methods (B3LYP) and the following 6-31G*, 6-31G** and 6-311G** basis set. The Gaussian program does semi-empirical and ab initio calculations.
We chose B3LYP as according to Monajjemi et al. (20) this method is appropriate for calculating NMR properties
Important information that can be gleaned from these calculations is the Hartree-Fock energy (ΔE), Atomic charge and Dipole moment (Debye). We used Gaussian98 at the NMR shift calculation using the B3LYP and 6-31G*, 6-31G** and 6-311G** basis set, as these calculations provide valuable information for exploring the experimental NMR chemical shifts. The chemical shielding refers to the phenomenon which associated with the secondary magnetic field created by the induced motions of the electrons that surrounding the nuclei when in the presence of an applied magnetic field for chemical shielding (CS) tensors, which describes how the size of shielding varies with molecular orientation. The two values of the shielding tensor are frequently expressed as the isotropic value (σiso), the anisotropy shielding (σaniso) and the other parameters (21,22).
The calculations also provide valuable information for exploring the thermodynamic parameters. Geometry optimization in NAA and AA was performed at the B3LYP method and using and 6-31G*, 6-31G** and 6-311G** basis set. We obtained the energy (ΔE), enthalpies (∆H), Gibbs free energy (∆G), entropies (∆S) of NAA and MAA (23,24).
We also studied electronic structures of NAA and MAA using Natural Bond Orbital (NBO) analysis at the same levels that were mentioned above in gas phase. A full NBO analysis is obtained in Gaussian 98 (25,26). The main listing of NBOs, displaying the form and occupancy of the complete set of NBOs that span the input AO space and for each orbital gives the type of orbital and the occupancy (27-29).
Result and Discussion
NMR parameters
In this section we report and analyze NMR shielding tensors of 1H, 13C, 17O-NMR such as isotropic shielding (σiso) and anisotropic shielding (σaniso) of MAA and its NAA, which obtain at B3LYP level using 6-31G*, 6-31G** and 6-311G** basis set in gas phase. The NMR technique is based on the sensitivity of magnetic properties. The calculation of NMR parameters using ab initio methods has important role in the molecular structure investigation. The quantitative knowledge of chemical shielding anisotropy (CSA) tensors is important in the context of bimolecular applications of nuclear magnetic resonance (NMR). In our current research, we have presented the results of our studies the intramolecular hydrogen bonding effects on the 1H, 13C, 15N-NMR shielding tensors of MAA and NAA. According to our theoretical data, it is apparent that the intramolecular hydrogen bonding effects seem quite significant. The 1H, 13C, 17O-NMR parameters of MAA and NAA are given in Table 1. According to Table 1, at three levels are shown the isotropic shielding value (σiso) and anisotropic shielding value (σaniso) for C2, C3, C4, O7, Oa and the hydrogen atom that in the intramolecular hydrogen bond formation is involved (H17 in NAA and H18 in MAA). The calculated results in Table 1 showed that isotropic shielding value (σiso) for H17 atom of NAA is smaller than H18 atom of MAA, while anisotropic shielding value (σaniso) for H17 atom of NAA is greater than H18 atom of MAA. In fact, H17 in NAA is more influenced by the magnetic field and is deshielder than H18. Also listed in Table 1 show that dipole moment of MAA is more than that NAA.
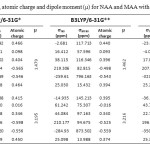 |
Table 1. NMR parameters, atomic charge and dipole moment (μ) for NAA and MAA with three levels. Click here to View table |
According to Table 1, in both structures the atomic charge of O7 atom is negative value whereas H17 and H18 atoms have positive values. Our obtained results from the analysis of the calculated values yielded strong evidence that existence of electron-pull and electron-donor substitute effect on the C3 atom and intramolecular hydrogen bonding effect play very important role in determining the 1H, 13C, 17O -NMR chemical shielding tensors of NAA and MAA. With B3LYP/6-31G* level, atomic charge O7 and H17 in NAA is -0.565 and 0.465 whereas atomic charge O7 and H18 in MAA is -0.598 and 0.450, respectively. Electronic effects plays important role in determining the chemical shielding tensors. The electron-donor substitutes increase electronic density and shielding value, while electron-pull substitutes decrease electronic density and shielding value. O7 in NAA has low atomic charge rather than O8 in MAA, there for H17 in NAA is free and contribute in formation of intramolecular hydrogen bonding. Also σiso value H17 is 26.060 ppm and H18 is 26.073 ppm. Our obtained results show good agreement was observed between atomic charges and NMR parameters.
Frequency calculations
The relative energy (ΔE), standard enthalpies (ΔH), entropies (ΔS), Gibbs free energy (ΔG) and constant volume molar heat capacity (Cv) values of NAA and MAA was obtained by theoretical methods using the 6-31G*, 6-31G** and 6-311G** basis set to obtain minima of the potential energy. In this paper according to values listed in Table 2, we compared intramolecular hydrogen bonding effect on thermochemical parameters of the NAA and MAA.
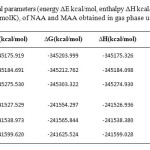 |
Table 2. Relative thermochemical parameters (energy ΔE kcal/mol, enthalpy ΔH kcal/mol, and Gibbs free energy ΔG kcal/mol, and entropy ΔS cal/ (molK), of NAA and MAA obtained in gas phase using three level Click here to View table |
The calculated results in Table 2 showed that the relative energy (ΔE) Gibbs free energy (ΔG) and standard enthalpies (ΔH) values of NAA in the three levels are negative values and entropy (ΔS) and constant volume molar heat capacity (Cv) values are positive. The thermochemical parameters values in NAA are larger than MAA that shows NAA is more stable than MAA.
Geometry optimization
Analysis of the geometrical parameters and according to values listed in Table 3 indicates that the main effect of 3-substitution is a lengthening of r (O7-H18) in MAA shortening of r(O7-H17) in NAA, that showing an electron-donor group decrease length bond O-H and electron-pull group increase length bond O-H. Also r(C2-C3) and r(C3-C4) in NAA is longer than MAA, while r(C2-O8) and r(C4-O7) in NAA is shorter than MAA. With existence electron-pull group and formation of intramolecular hydrogen bonding increase length bond O-H but reduce length bond C-O.
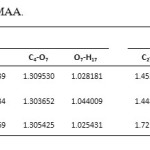 |
Table 3. Bond lengths ( r, A ͦ ) for NAA and MAA. Click here to View table |
NBO analysis
Natural bond orbital analysis provides the accurate possible natural Lewis structure. The result of interaction is a loss of occupancy from the concentration of electron NBO of the idealized Lewis structure into an empty non-Lewis orbital. A careful examination of all possible interactions between ‘‘filled’’ (donor) Lewis-type NBOs and ‘‘empty’’ (acceptor) non-Lewis NBOs, allows us to get an estimate of their energetic importance by second-order perturbation theory. For each donor (i) and acceptor (j), the stabilization energy E(2) associates with the delocalization i j. The strengths of these delocalization interactions, E(2), are estimated by second order perturbation theory. Some of significant donor–acceptor interactions and their second order perturbation stabilization energies E(2) which were NAA and MAA are given in Table 4. This section shows some of the donor–acceptor
interactions and their second order perturbation energies (E(2)) for NAA and MAA.
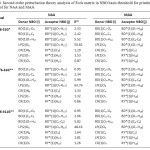 |
Table 4. Second order perturbation theory analysis of Fock matrix in NBO basis threshold for printing: 0.50 kcal/mol for NAA and MAA Click here to View table |
It seems that in NBO analysis of hydrogen bond systems, the charge transfer between the lone pairs of proton acceptor and antibonds of proton donor is most significant. The results of the NBO analysis in Table 4 show that in NAA structure, LP(2)O8 participates as donor and the BD*(O7–H17) interactions as acceptor in intramolecular hydrogen bonding interaction and in MAA structure, LP(2)O8 participates as donor and the BD*(O7–H18) interactions as acceptor in intramolecular hydrogen bonding interaction. Electron density is transferred from one pair LP(2)O8 to the anti-bonding BD*(O7–H17) orbital’s in NAA and LP(2)O8 to the anti-bonding BD*(O7–H18) orbital’s in MAA. The resonance energy (E(2)) indicates amount of Participation of electrons in the resonance.
According to the simple bond orbital picture, each bonding NBO is defined as an orbital formed from two directed valence hybrids (NHOs) hA, hB on atoms A and B, with corresponding polarization coefficients cA, cb. Table 5 show share of orbitals contribute in the bonds (BD for 2-center bond). According to Table 3, by using B3LYP/6-311G** for NAA, in the O7-H17 bond, BD= 0.9052sp2.71d0.01 + 0.4250s reported. Polarization coefficients of the O7-H17 bond O7= 0.9052 and H17= 0.4250 reported, that sizes of these coefficients show the importance of the hybrid of O7 in the formation of the bond, while for MAA in the O7-H18 bond BD= 0.8990sp2.73d0.01 + 0.4379s reported. Polarization coefficients of the O7-H18 bond O7= 0.8990 and H18= 0.4379 reported, that sizes of these coefficients show the importance of the hybrid of O7 in the formation of the bond. Also values of Polarization coefficients H17 and H18 show share of contribute H18 in bond O7-H18 at MAA is greater than
share of contribute H17 in bond O7-H17 at NAA. There for H17 in NAA greater than H18 in
MAA contribute in formation of intramolecular hydrogen bonding.
 |
Table 5. Calculated NHOs and the polarization coefficient for each hybrid in the corresponding NBO (in parentheses) for NAA and MAA. Click here to View table |
The last section allows to quickly identify the principal delocalizing acceptor orbitals associated with each donor NBO, and their topological relationship to this NBO (see Table 6). These data show amount of occupancy and energy of each orbital. According to Table 6, seems that LP(2)O8 (donor NBO) in NAA and MAA is lowest orbital occupanced and has highest energy that delocalizing acceptor orbitals in NAA [BD*(1)O7-H17] and MAA [BD*(1)O7-H18].
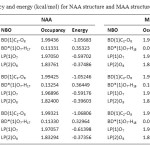 |
Table 6. Occupancy and energy (kcal/mol) for NAA structure and MAA structure. Click here to View table |
In the present study, we used a combination of theoretical tools to compare nitroacetylacetone (NAA) structure and methylacetylacetone (MAA) structure. The following conclusions are obtained from the current study:
1. The most stable structure, according to the optimization energy is NAA. It seems that intramolecular hydrogen bonding in NAA is stronger than MAA.
2. NBO analysis indicated the presence of donor-acceptor centers in the investigated structures. In both the structures the resonance energy (E(2)) indicates amount of Participation of electrons in the resonance. The comparison between the NBO analysis of two Compounds shows that values of E(2) for MAA are lower than NAA which means that in the MAA structure lesser electrons are involved in the resonance.
3. The σiso value of H17 in the NAA structure is lower than σiso value of H18 in MAA structure. This means that electron density around H17 is lower than H18. There for participation of H17 at formation intramolecular hydrogen bonding is higher than that H18 in MAA.
4. The comparison of thermochemistry parameters of two structures that are expressed show that ΔG, ΔH, ΔE, ΔS and Cv values for NAA are more than those of MAA which again indicates the greater stability of NAA.
5. Analysis of the geometrical parameters and according to values listed showing an electron-donor group decrease length bond O-H and electron-pull group increase length bond O-H. With existence electron-pull group in NAA and formation of intramolecular hydrogen bonding increase length bond O-H but reduce length bond C-O.
6. Finally, our studies on the structures showed that intramolecular hydrogen bonding in NAA stronger than that MAA and NAA structure and NAA is more stable than MAA structure.
References
- Schuster P., Zundel, G. and Sandorfy C., The Hydrogen Bond, vol. 1-3, North Holland Publ. Co, Amsterdam, (1976).
- Buckingham A. D, Powler, P. W. and Hutson, J. M., Chem. Rev., 88, 963(1988).
- Ferreiro M. L. and Rodriguez-Otero J., J. Mol. Struct. THEOCHEM., 542, 63 (2001).
- Buemi G. and Zuccarello F., Chem. Phys., 306, 115(2004).
- Tayyari S.F, Raissi H, Milani-Nejad F. and Butler I.S., Vib. Spectrosc., 26, 187(2001).
- Raissi H. F, Farzad F. and Nowroozi A., J. Mol. Struct., 752, 130(1972).
- Andreassen A. L. and Bauer, S.I., J. Mol. Struct., 12, 381(1972).
- Tayyari S.F. and Milani-Nejad, F., Spectrochim. Acta., 56, 2679(2000).
- Emsley. J., Struct. Bonding, Vol. 57, Springer, Berlin, 147(1984).
- Hibbert F. and Emsley J., Hydrogen Bonding and Chemical Reactivity; Advances in Physical Organic Chemistry, Vol. 26, Academic Press, London, 255(1990).
- Emsley J., Structure and Bonding, Vol. 57, Springer, Berlin, 147(1984).
- Yoshida Z, Ogoshi H. and Tokumitsu, T., Tetrahedron., 26, 5691(1970).
- Emsley J, Freeman N. J, Bates P. A. and Hursthouse, M. B., J. Chem. Soc. Perkin Trans., 1, 297(1988).
- Emsley J, Ma L. Y. Y, Bates P. A. and Hursthouse M. B., J. Mol. Struct., 178,297(1988).
- Emsley J, Ma L. Y. Y, Bates P. A, Motevalli M. and Hursthouse M. B., J. Chem. Soc. Perkin Trans., 2, 527(1989).
- Emsley J, Ma L. Y. Y, Karaulov S. A, Motevalli M. and Hursthouse M. B., J. Mol. Struct., 216, 143(1990).
- Emsley J, Ma L. Y. Y, Nyburg S. C. and Parkins A.W., J. Mol. Struct., 240, 59(1990).
- Buemi G. and Zuccarello F., J. Theor. Ch., 2, 302(1977).
- Becke A. D., Phys. Rev. A., 38, 3098(1988).
- Monajjemi M, Afsharnezhad S, Jaafari M. R, Abdolahi T, Nikosade A, Monajemi H.,Russ. J. Phys. Chem. A., 81, 1956(2007).
- Monajjemi M, Azad M. T, Haeri H. H, Zare K, Hmedani Sh., J. Chem. Res., 8, 454(2003).
- Monajjemi M, Sheikhi M., J. Phys. Theor. Chem. IAU Iran., 8, 119(2011).
- Monajjemi M, Afsharnezhad S, Jaafari M. R, Abdolahi T, Nikosade A, Monajjemi H.,Phys. Chem. Liquids., 49, 318(2011).
- Monajjemi M, Mahdavian L, Mollaamin F, Honarparvar B., Fullerenes, Nanotubes Carbon Nano-structures., 18, 45(2010).
- Monajjemi M, Sheikhi M, Mahmodi Hashemi1 M, Molaamin F. and Zhiani R., International Journal of Physical Sciences., 7, 2010(2012).
- Gnanasambandan T, Gunasekaran S. and Seshadri S., Orient. J. Chem., 29, 185(2013).
- Noei M, Salari A. A, Baei M. Hajizadeh T. F, Kazemi Asl, J, Ramezani M., Arabian Journal of Chemistry., 2011.
- Scholtzova E, Mach P, Langer V., Acta Cryst. A., 65, 270 (2009).
- Heidarnezhad Z, Ganiev I, Obidov Z, Heidarnezhad F. and Sharifi M. S., Orient J. Chem., 28, 1597(2012).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


