Influence of Hydration on the Neutral Complex (-OH, H3O+) (H2O)n
F. Benyettou1*, S. Hiadsi2 and A. Bendraoua1
1Department of Chemistry, University of Mohamed Boudiaf, USTO -BP 1505 El Mnaouer, Oran, Algeria.
2Department of Physique, University of Mohamed Boudiaf, USTO -BP 1505 El Mnaouer,Oran, Algérie.
Corresponding Author E-mail: leau_lautoprotolyse@yahoo.fr
Density-functional theory (DFT) within local density approximation (LDA) has been carried out for a sequence of hydrated proton clusters. Optimised structures were odtained for n=0, 1 and 3 for (-OH, H3O+)(H2O)n, complex . Hydronium ion H3O+ and hydroxide ion OH- are found to be the center of the neutral complex (-OH, H3O+). The LDA give shorter hydrogen bond lengths O-H, but slightly longer chemical bond lengths O…H as compared with the Post-Hartree-Fock calculations. We found that the distance O...H successively increases with the number of water molecules added to the neutral complex (-OH, H3O+). The solvent effects on proton transfer energy barriers in clusters have been studied. The harmonic vibrational frequencies and IR intensities of various modes have been generated for all optimised structures. This study was confirmed by vibrational studies of these complexes, our results give excellent agreement with experimental values.
KEYWORDS:hydrated proton clusters; density functional theory; the proton transfer; ab initio methode; intermolecular interactions; hydrogen bond
Download this article as:| Copy the following to cite this article: Benyettou F, Hiadsi S, Bendraoua A. Influence of Hydration on the Neutral Complex (-OH, H3O+) (H2O)n. Orient J Chem 2013;29(2). |
| Copy the following to cite this URL: Benyettou F, Hiadsi S, Bendraoua A. Influence of Hydration on the Neutral Complex (-OH, H3O+) (H2O)n. Orient J Chem 2013;29(2). Available from: http://www.orientjchem.org/?p=22066 |
Introduction
The state of a proton in aqueous solution and clusters is of wide interest and the proton Transfer reaction is one of the most important in chemistry [1]. Proton transfer also plays a very important role in biological processes [2-5], however a proper description of such a process is still a major challenge for theoretical chemistry. Advances in theory and the availability of increased computer power have allowed substantial progress to be made for proton transfers in solution [6-8] and clusters [9,10].
In recent months, the methods and techniques of density functional theory(DFT) have advanced remarkably[11].They show considerable advantages of generality and accuracy with respect to the semiempirical methods and are more rapid than the highly correlated traditional ab initio methods that are required to treat questions of reactivity. They have been used to study small hydrated proton clusters H2O+5 [12,13] and other hydrogen-bonded systems [14]. The energy barrier to proton transfer was calculated using [12] and compared with the results of other computational methods. A combined ab initio density functional and classical molecular dynamics simulation (DFT CDFMD) [15] is under development to study quantum molecules in solution. It can be applied, in its present state, to calculate the free energy barriers of proton transfer in solution.
Small clusters provide a unique solvent environment, where reaction dynamics can be very different from that in aqueous solution. Recent advances in laser technology and molecular beam spectroscopy allow experimentalists to provide detailed information on the structure of the cluster. High resolution vibration-rotation spectroscopy of the hydrated hydronium ions, e.g., H5O+2, H7O+3 and H9O+4 have been obtained by lee’s group [16] .Many ab initio calculations have been published for H5O+2 [9,10,12]. However, to our knowledge, except for some earlier results of Newton [17] and unpublished works of Remington and Schaefer [18], very few ab initio results are available for the optimised structures and vibrational analyses for larger hydrated proton clusters.
In this paper, we report the ground state structures and the vibrational analysis for the hydrated proton the neutral complex (–OH, H3O+)(H2O)n.lusters with n=0,1and 3. Few of the method used in ab initio are designed to probe the solvent effects on the energy barriers of proton transfer in the hydrated proton the neutral complex.
The aim of our research is to study the effect of the interaction of several bodies on the structure of complex systems in the neutral (–OH, H3O+)(H2O)n, with (n = 0, 1, 3). These systems represent great interest in a chemical point of view in the biological field.
The study of these systems, it also provides information on the structure, the number of coordinate hydration and the nature of the routesto from neutral complexes.
Our job is to study the properties of aqueous ionic solutions by ab initio calculations on the systems:(–OH, H3O+) with symmetry C1, (–OH, H3O+)(H2O) with symmetry C1, (–OH, H3O+)(H2O)3, with symmetry Cs. We will discuss three methods: Hatree-Fok, Moller Plesset perturbation, DFT with the 6-31 + G basis.
The remainder of this article is divided into three parts. The computational method is discussed in Sec.II, results are presented in Sec.III, and our conclusions are briefly summarized in Sec.IV.
II.Computational Methods
We have computed the optimal geometries and harmonic vibrational frequencies of complex systems in the neutral (–OH, H3O+)(H2O)n, with (n = 0, 1, 3).A geometry was considered as optimised when the gradient was less than 0.0001 a.u.
All calculations were carried out in the ab initio [19] molecular orbital ( MO) framework, using basis sets of contracted Gaussian-type orbitals (GTO’s). The intergral and SCF programs employed are those recently developed by Hehre and Pople [20] .Wedesired a basis set sufficiently flexible to give reasonable quantitative account of the structure and intermolecular energitics of the system under study while at the same time small enough so that a detailed study of potential energy surfaces of interest might be carried out economically. In preliminary minimal-basis calculations 6-31 + G ** [27] This includes polarization functions on all atoms
with different levels; coerenti self-field Hartree-Fock (HF) [21], the perturbation method of Moller-Plesset (MP) to second order [28-29]., the density functional method (DFT) [22-26] calculations with the perdew nonlocal corrections to exchange and correlation give somewhat shorter hydrogen bond lengths.
III.Results And Discussion
We studied the influence of hydration of the neutral complex (–OH, H3O+)(H2O)n, by ab nitio’s calculation ,with structural, vibrational ,and energitical point of view. We started our study with the complex (–OH, H3O+) which has a symmetry with the C1 complex then (–OH, H3O+)(H2O) with a C1 symmetry and the complex(–OH, H3O+)(H2O)3 which has a Cs symmetry, this study has been addressed by methods HF, MP2 and DFT / B3LYP with the 6-31 + G **, all results are grouped in Tables (1,2,3,4).
The intra-and intrermolecular coordinates complex (–OH, H3O+) were optimized by three methods is (Table.1) . The system stabilizes at a level of energy – 95426.04 kcal / mol by the HF method, we notes in (Fig.1). The donor monomer ion H3O+ has three covalent bonds, two bonds among them in the same plans, those formed by the oxygen atom O1 and the hydrogen atoms H4, H2 which has a distance O1-H4 and O1-H2 equivalent to 0.944 Å. The route that is outside of the plan is formed by the O1 oxygen atom and the hydrogen atom H3 at a distance O1-H3 of 0.963 Å. The angle O1-H3-H4 is 117.1 °, the angle of H3-O1-H2 is 114.8 °. °H2-H4-O1 is 107.1 °, these three angles are closed to the angle 120°.
The donor monomer combined with the acceptor monomer–OH by a O5…H4 hydrogen at a distance of 2.001 Å , which confirms that the three atoms O1, O5 and H4 are in the same plane almost linear. A comparison of results with the values found in experimental observation hat the HF method is close to the experimental 0.04008% compared to the DFT method 0.04358% and the MP2 method 0.04226%.
The vibrational study of complexes with different methods HF, MP2 and DFT was made by the 6-31 + G ** basis, see (Table.2).
This table has been divided into two parts the first is the intermolecular vibration and the second intermolecular vibration. To explain this bungs have chosen the HF method, the appearance of three frequency bungs (137-630, 1743-1758, 4096-4257)Cm-1 of varying intensities (Table.2). The two first bungs correspond to intermolecular vibrations and the third bung corresponds to the intermolecular vibrations, the frequency bung (4096-4257) Cm–, reply the OH stretch vibration, the band (1743-1758) Cm-1 is consistent with the deformation vibration (Bending), and the bung (137-630) Cm-1 corresponds to the intermolecular vibrations of the OH bridge linking hydrogen
]. O-H….. O]. A comparison with experimental values 3385 and 3490 Cm-1 reflects the values of frequencies calculated (Table2).
The next step, we add to this complex a water molecule to see the influence of the latter on the neutral complex (–OH, H3O+). The system (–OH, H3O+)(H2O) stabilizes with C1 symmetry, see (Fig. 2) at an energy level -143147.7 k cal / mol for the HF method see (Table 3). A comparison of results found with the experimental observation that in the HF method is closed to the experimental 0.04008 % compared to the DFT method 0.04358 % and the MP2 method 0.04226 %[30]. We noticed that there’s a decrease of O-O distance of about 0.12 Å, and hydrogen bond distance of about 0.19 Å, there also a significant increase in binding of the –,OH ion H3O + with a value of approximately 0.035 Å, and an increase in the angle of the ion of about 6 °even (Table 3).
The calculation of frequencies of vibration in these complexes restores the bung in the same frequency range, but with different intensities (Table.4) and the bung (277-870) Cm-1 corresponds to the intermolecular vibrations of the OH bridge linking hydrogen. O-H …..O., three (648,401, 617, 83, 583, 861) Cm-1 reply to the three hydrogen bonds of the appeared complex. By comparison with experimental values 3385 and 3490 Cm-1 . We deduct, that the experimental values correlate well with the frequency values calculated for different levels in (Table.4). We recently added to the neutral complex (–OH, H3O+) three water molecule to study the intra-and intermolecular interaction of the system(–OH, H3O+)(H2O)3 , with an even symmetry Cs (Fig .3), an energy level – 95933.46 k cal / mol for the HF method see (Table 3).The results are shown in Table 3 and Tables .4., A comparison with results found in the experimental observation ,also we notice that the DFT method is closed to the experimental 0. 0152 % compared to the HF method 0. 0215 % and the MP2 method of 0. 0243 % . By varying the coordinat number there was a slight increase in distance R O-O 0. 02 Å with a slight increase in hydrogen bonding distance of 0. 21 Å. There is a decrease of O-H bond in relation to the first with a value of 0. 01 Å, with a decrease of angle to the first with a value of 2 °. even .1,2,3 figures.
The calculation of frequencies of vibration in these complexes, gives a band (181-840)
Cm-1corresponds to the intermolecular vibrations of the O-H bridge linking hydrogen
O-H …..O six frequencies (534.352- 566.166- 608.95- 696.731-811.627-833.038) Cm-1 responds to the six hydrogen bonds of appeared complex. and other bungs frequencies in the same interval but with different intensities (Tableau.4) that can be explained by the number of coordinat hydration and their positions (the symmetry of the complex), simply the influence of hydration has a great interest on the structure of the complex (–OH, H3O+) in comparison with experimental values 2700 Cm-1 , 3025 Cm-1 , 2600 Cm-1 [33] and 2800 Cm-1, it is deduced that the experimental values correlate well with the values calculated for different frequency levels.
IV. Conclusion
A comparative structural and intra-and intermolecular energy by different levels MP2, HF, DFT with 6-31 + G ** basis on the neutral system (–OH, H3O+)(H2O)n. The analysis of results has allowed us: to determine the configurations of coordinates that represent the complex neutral (–OH, H3O+)(H2O)n. We notice that there is an important bonding increase of the –OH, H3O+ by adding water molecule in the complexe. In the comparative study of the hydrogen bond distance of a monomer to monomer donor acceptor complex in the neutral, we find that the distance O….H was reduced by the addition of three water molecules in complex and the energy gets weaker more and more by the addition of water molecule to the complex, it is predicted by the frequency of vibration.
We can conclude that this study has provided information on the structure, number of coordinat hydration and nature of the neutral complexes bonds, and our results give an excellent agreement with experimental values.
Table 1: The geometric parameters calculated at different levels of (–OH, H3O+) and the comparison between results from experimental values.
|
Internal Coordinate |
HF |
MP2 |
DFT |
Experiment a |
|
R(O(1) -H(4)), [Å] R(O(1) -H(2)), [Å] R(O(1) -H(3)), [Å] y(H(3)-O(1)-H(2)),°. y(H(1)-O(4)-H(5)),°. y(H(3)-O(1)-H(4),°. y(H(2)-O(1)-H(4)),°. R(O(5) -H(6)), [Å] R(O(5) -H(4)), [Å] |
0. 94417 0. 94423 0.96353 114. 8552 55.9870 117. 13855 107. 13855 0. 94244 2. 001234 |
0. 9643 0. 9643 0, 97561 116. 0936 55. 2015 115. 2018 105. 679 0. 9624 2. 009184 |
0. 9658 0. 9658 0. 965859 113. 1782 53. 5312 112. 3741 106. 0589 0. 9642 1. 923828 |
0. 950 0. 950 0. 950 111. 2 – 111. 2 104.5 0. 950 – |
aReference 30
Table 2 : Harmonic vibration frequencies (in Cm -1) in the complex (–OH, H3O+) and experimental values the comparison between results from previous and experimental values.
|
|
System |
MP2 (cm -1 ) |
MP2 IR Intensities (km/mol) |
HF (cm -1 ) |
HF IR Intensities (km/mol) |
DFT (cm -1 ) |
DFT IR Intensities (km/mol) |
Expriment a (cm -1 ) |
|
Intermolecular |
|
|
|
|
|
|
|
|
|
ω9(a”) |
Acceptor asymmetric O-H Stretch |
4000. 68
|
95. 8597
|
4257. 29
|
121. 565 |
3924. 24 |
86. 8857 |
|
|
ω1(a’) |
Donor free O-H Stretch |
3978. 55
|
124. 758
|
4244. 24
|
146. 707
|
3898. 78
|
95. 0156
|
3490
|
|
ω2(a’) |
Acceptor symmetric O-H Stretch |
3861. 32
|
14. 244
|
4141. 42
|
29.0664
|
3808. 5
|
13. 9001
|
3385
|
|
ω3(a’) |
Donor bridge H Stretch |
3788. 01
|
260. 284
|
4096. 08
|
213. 66
|
3696. 96
|
332. 356
|
|
|
ω4(a’) |
H3O+ donor bend |
1658.37 |
64.9643 |
1758. 94 |
92.7181 |
1631. 95 |
46.8163 |
|
|
ω5(a’) |
–OH acceptor bend |
1636.92 |
109. 655 |
1743. 95 |
135. 431 |
1616. 03 |
123. 793 |
|
|
ω10(a”) |
H..(H-O-H) Out of plane donor bend |
676.401 |
192. 918 |
630.804 |
232. 658 |
681.336 |
171. 814 |
|
|
ω6(a’) |
In- plane donor wag |
372. 019 |
128. 61 |
359. 79 |
140. 984 |
393. 602 |
97.2357 |
|
|
ω7(a’) |
O-O stretch |
202. 215 |
172. 523 |
186. 384 |
248. 51 |
205. 659 |
198. 959 |
|
|
ω8(a’) |
Acceptor bend |
178. 67 |
67.7546 |
167. 948 |
78. 2911 |
179. 521 |
35. 652 |
|
|
ω11(a”) |
Acceptor twist
|
170. 78
|
129. 816
|
157. 579
|
87 . 4746
|
170. 518
|
128. 775
|
|
|
ω12(a”) |
Donor torsion |
144. 827 |
143. 051 |
137. 783 |
136. 708 |
144. 128 |
182. 66 |
Table 3: internal study structure of the coordinate by taking into account the inter and intra-molecular interactions on the studied complexes .
|
|
HF |
|
|
MP2 |
|
|
DFT |
|
Experiment |
|
|
|
n= 0 |
n=1 |
n=3 |
n= 0 |
n=1 |
n=3 |
n= 0 |
n=1 |
n=1 |
|
|
Intermolecular |
||||||||||
|
R(O-O) [Å]
|
2. 83405 |
2. 8872 |
2. 90 392 |
2. 91435 |
2.79677 |
2. 80547 |
2. 889690 |
2. 78975 |
2.785082 |
– |
|
R(O…H-O) [Å] |
2. 00123 |
2. 0341 |
2. 04913 |
2. 00918 |
1. 9194 |
2. 03678 |
1. 923828 |
1. 8834 |
2. 040007 |
– |
|
a y(H-O…H) |
55. 9870 |
148. 3984 |
107. 637 |
55. 2015 |
147. 363 |
107. 574 |
53. 5312 |
149. 38 |
107. 9534 |
104.5 a |
|
b y(H-O-H) ° |
117. 1385 |
91. 679 |
98. 815 |
115. 201 |
91. 6705 |
92. 3665 |
112. 3741 |
90. 951 |
97.15347 |
104.5 a |
|
107. 1385 |
107. 588 |
107. 869 |
105. 679 |
106. 476 |
106.714 |
106. 0589 |
106. 914 |
106. 951 |
111.2 a |
|
|
114. 8552 |
129. 6005 |
132. 895 |
116. 093 |
127. 810 |
131. 338 |
113. 1782 |
124. 22 |
130. 548 |
– |
|
|
R(O-H)c [Å] |
0..94417 |
0. 9508 |
0. 959813 |
0. 9643 |
0. 97561 |
0. 98345 |
0. 9658 |
0. 9801 |
0.985645 |
0. 950 a |
|
0. 94423 |
0. 9427 |
0. 960 |
0. 9643 |
0. 97561 |
0. 963 |
0. 9658 |
0. 9643 |
0. 972 |
1. 017± 0.005b |
|
|
0. 96353 |
0. 9589 |
0. 97123 |
0. 97561 |
0. 9612 |
0. 9749 |
0. 96586 |
0. 983 |
0. 984084 |
1. 01± 0.01c |
|
|
R(O-H)d [Å] |
0. 94244 |
0. 9424 |
0. 96201 |
0. 9624 |
0. 9627 |
0. 9758 |
0. 9642 |
0. 964 |
0. 964 |
0. 964 a |
a Angle antre les deux ions y(O-H…O)
b Angle de lion H3O+
c Distance de lion H3O+
d Distance de lion OH–
aReference 30
bReference 31
cReference 32
Table 4: Harmonic vibration frequencies (in Cm -1) in the complex undermentioned and comparison between the experimental values.
|
System |
MP2 (cm -1 ) |
MP2 IR Intensities (km/mol) |
HF (cm -1 ) |
HF IR Intensities (km/mol) |
DFT (cm -1 ) |
DFT IR Intensities (km/mol) |
Expriment (cm -1 ) |
|
(–OH, H3O+)( H2O)
|
3969, 37 |
121, 161 |
4426, 65 |
946, 269 |
3899.19 |
57.2785 |
3490 |
|
3967, 98 |
111, 378 |
3980, 03 |
140, 362 |
3898.01 |
106.036 |
3385 |
|
|
3964, 63 |
110, 833 |
3945, 8 |
27, 3588 |
3893.22 |
97.4185 |
||
|
3730, 38 |
405, 507 |
3942, 27 |
93, 1109 |
3616.28 |
541.255 |
||
|
3722, 19 |
444, 006 |
3677, 15 |
633, 836 |
3603.68 |
596.1 |
||
|
1653, 42 |
116, 771 |
1828, 27 |
196, 862 |
1631.08 |
111.833 |
||
|
1649, 01 |
132, 572 |
1757, 69 |
74, 0879 |
1626.48 |
117.6 |
||
|
861.006 |
19.3183 |
915, 145 |
317, 613 |
912.853 |
16.1281 |
||
|
700, 657 |
455, 086 |
834, 851 |
319, 188 |
722.42 |
428.2 |
||
|
565, 813 |
179, 755 |
719, 518 |
53, 503 |
604.778 |
213.997 |
||
|
347, 639 |
124, 807 |
648.401 |
147.529 |
473.567 |
153.197 |
||
|
259, 698 |
42, 5834 |
617, 83 |
126, 958 |
380,.945 |
50.2188 |
||
|
210, 193 |
180, 261 |
583, 861 |
199, 244 |
270, 584 |
32, 195 |
||
|
192, 581 |
12, 1642 |
371, 754 |
55, 4202 |
250, 727 |
64, 5898 |
||
|
451, 07 |
150, 232 |
276, 676 |
8, 2816 |
177, 446 |
120, 171 |
||
|
(–OH, H3O+)(H2O)3 |
3775,99 |
693,479 |
423994 |
191,149 |
3896.98 |
60.5471 |
3025 |
|
3722,56 |
143,642 |
4237,5 |
189,00 |
3895.4 |
130.992 |
2800 |
|
|
3712,24 |
627,636 |
4127,91 |
517,08 |
3894.05 |
126.164 |
2700a |
|
|
3708,41 |
184,877 |
4063,3 |
453,999 |
3644.82 |
832.215 |
2600a |
|
|
3667,24 |
287,406 |
4036,96 |
199,898 |
3597.31 |
535.396 |
||
|
1651,57 |
143,642 |
1790,09 |
22,4773 |
3592.66 |
965.33 |
||
|
1648,81 |
115,974 |
1751,9 |
174,096 |
1431,06 |
186,177 |
||
|
897.157 |
29.0476 |
833.038 |
106.803 |
779.342 |
439.353 |
||
|
760.545 |
412.5 |
811.627 |
4.2433 |
690.921 |
32.6021 |
||
|
668.238 |
417.687 |
696.731 |
512.902 |
670.105 |
288.214 |
||
|
641.291 |
24.0444 |
608.95 |
461.502 |
633..506 |
155.281 |
||
|
598.752 |
267.709 |
566.166 |
19.4118 |
572.159 |
161.968 |
||
|
404,899 |
115.238 |
534.352 |
263.968 |
430.787 |
25,2856 |
||
|
380,691 |
162,138 |
473,519 |
142,43 |
416,043 |
145,337 |
||
|
213,472 |
122,684 |
366,969 |
137,189 |
386,379 |
386.622 |
||
|
226,801 |
165,693 |
205,617 |
70,304 |
270, 584 |
13,8428 |
||
|
193,942 |
144,634 |
237,448 |
11,1279 |
250, 727 |
32, 195 |
||
|
180,813 |
67,7975 |
198,492 |
21,3725 |
aReference 33.
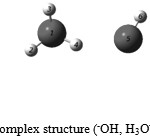 |
Figure 1: The neutral complex structure (–OH, H3O+) with C1 symmetry.: Click here to View figure |
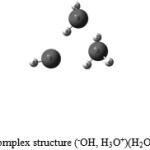 |
Figure 2: The neutral complex structure (–OH, H3O+)(H2O) with C1 symmetry.: Click here to View figure |
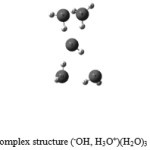 |
Figure 3: The neutral complex structure (–OH, H3O+)(H2O)3 with CS symmetry.: Click here to View figure |
References
- G.W.Robinson, P.J.Thistlethwaite, and J.Lee,J.Phys.Chem.90.4224(1986).
- E.N.Baker and R.E.Hubbard, Prog.Biophys.Mol.Biol, 44,97(1984).
- G.A.Jeffrey and W.Saengeri, in Hydrogen Bonding in Biological Structures (Springer, New York,.1991).
- Y.Cha,C.J.Murray, and J.P.Klinman, Science 243, 1325 (1989);J.Rucker, Y.Cha, T.JONSSON, K.L.Grant, and J.P.Klinman, Biochemistry 31, 11489 (1992).
- A.Warshel,Computer Modelling of Chemical Reactions in Enzymes and Solutions (Wiley, New York, (1991)
- D.Borgis, S .Lee, and J.T.Hynes, Chem.Phys. Lett.19,162 (1989); D.Borgis, G.Tarjus, and H.Azzouz, J.Phys. Chem;96,3188 (1992); D.Li and G.Voth, ibid, 95,10425 (1991).
- D.Borgis and J.T.Hynes, J.Chem.Phys. 94, 3619 (1991).
- D.Laria, G.Ciccotti, M.Ferrario, and R. Kapral, J.chem. Phys. 97, 378 (1992);J.Lobaugh and G.A.Voth, Chem.Phys. Lett. 198, 311 (1992).
- S.Scheiner, Acc.Chem.Res.18, 174 (1985).
- K.Luth and S.Scheiner, J.Chem.Phys.97, 7507 (1992); Z.Latajka and S.Scheiner, J.Mol.Struct. 234, 373 (1991).
- D.R.Salahub, M.A.Castro, and A.Proynov, in Relativistic and Electron Correlation Effects in Molecules and Solids, edited by G.L.Malli (Plenum, New York, 1994).
- C.Mijoule, Z.Latajka, and D.Borgis, Chem.phys.Lett. 208, 364 (1993).
- Santhanamoorthi Nachimuthu, Jiali Gao, Donald G. Truhlar Chemical Physics 400 (2012) 8–12
- F.Sim, A St-Amant, I.Papai, and D.R.Salahub, J.Am.Chem.Soc 114, 4391 (1992).
- D.Q.Wei and D.R.Salahub, Chem. Phys.Lett. 224, 291 (1994).
- L.I.Yeh, M.Okumura, J.D.Myers,J.M.Price, and Y.T.Lee, J.Chem.Phys 91, 7319 (1989).
- M.D.Newton, J.Chem.Phys.67, 5535 (1977).
- R.Remington and H.F.Schaefer (unpublished results) quoted in Ref.15.
- David. Tozer, Chengteh Lee and George Fitzgerald, J.Chem.Phys.104, 5555 (1996).
- Selinger and A.W.Castleman. Jr, J.Phys.Chem.95,8442(1991).
- C.Moller and M.S.Plesset, Phys.Rev.46,618 (1934).
- W.Kohn and L.J.Sham, Phys. Rev.A.140,A1133 (1965).
- Density Functional Methods in Chemistry, edited by J.K.Labanowski and J.W.Andzelm (Springer-Verlag, New York, (1991).
- F.Sim.A. St. Amant.J.Papai, and D.R.Salahub, J.Am.Chem.Soc.114,4391 (1992).
- H.B.Schlegel,J.Comput,Chem.3,214(1982).
- Gaussian98. Revision.M.J.Frisch,G.W.Trucks,II.B.Schlegel.G.E.Scusecria,M.A.Robb, J.R.Cheeseman,V.G.Zakrzewski,J.A. Montgomery, Jr.r.e.Stratmann, J.C.Burant, S.Dapprich, J.Millam,A.D.Daniels, K.N.Kudin, M.C.Strain, O.Farkas, J.Tomasi,V.Barone,M.Cossi,R.Cammi, B.Mennucci, C.Pomelli, C.Adamo, S.Clifford, J.Ochterski, G.A.Pertersson, P.Y.Ayala, Q.Cui, K.Morokuma,D.K.Malick,A.D.Raduck,K.Raghavachari,J.B.Forestman,J.Croslowski,J.V.Ortiz,B.B.B.Stetanov,G.Liu,A.Liashenko,P.Piskorz,I.Komaromi,R.Gomperts,R.L.Martin,D.J.Fox,T.Keith,M.A.AI-Laham, C.Y.Peng,A.Nanayakarra,C.Gonzalez, M.Challacombe,P.M.W.Gill, B.Johnson,W.Chen,M .W.Wong,J.L.Andres,L.Gonzalez,M.Head-Gordon,F.S.Replogle and J.Apople,Gaussian,Inc ,Pittsburgh PA ,(1198).
- J.A.Pople, R.Seeger, and R.Krishnan, Int.J.Quantum Chem.Symp.11149 (1977)14.
- R.Krishnan and J.A.Pople, Int.J.Quantum. Chem.14, 91 (1978).
- R.Krishnan, M.J.Frisch, and J.A.Pople, J.Chem.Phys.72,4244(1980).
- K.Kuchitsu and Y.Morino, Bull.Chem.Soc. Japan. 38, 814 (1965).
- J.O.Lundgren and I.Olovsson, J.Chem.Phys.49, 1068(1968).
- J.O.Lundgren and J.M.Williams, J.Chem.Phys.58, 788 (1973).
- P.A.Giguére and S.Turrell, Can.J.Chem.54, 3477(1976).,Inspec, this work is a followup of an earlier, pioneering study by M.Falk and P.A.Giguére, Can.J.Chem.35, 1195(1957).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


