2D-QSAR Assisted Design, and Molecular Docking of Novel Indole Derivates as Anti-Cancer Agents
Meenakshi Rana1 , Niladry Sekhar Ghosh2*, Dharmendra Kumar3, Ranjit Singh1and Jyoti Monga4
, Niladry Sekhar Ghosh2*, Dharmendra Kumar3, Ranjit Singh1and Jyoti Monga4
1Department of pharmaceutical sciences, Shobhit University, Gangoh, Saharanpur UP. India
2Faculty of Pharmaceutical Sciences, Assam down town University, Guwahati, Assam. India
3Narayan Institute of Pharmacy. Gopal Narayan Singh University,Jamuhar, Sasaram, Bihar. India
4Ch. Devi Lal College of Pharmacy, Jagadhri, Haryana, India
Corresponding Author E-mail:niladry_chem@yahoo.co.in
DOI : http://dx.doi.org/10.13005/ojc/400527
Article Received on : 11 Jun 2024
Article Accepted on : 01 Oct 2024
Article Published : 06 Nov 2024
Reviewed by: Dr. Parameshwara Naik P
Second Review by: Dr. Naresh Batham
Final Approval by: Dr. Bal Krishan Sharma
CADD is an important aspect of the any currently employed drug discovery process for a medicinal chemist. In the current study, research was initiated with a two dimensional Quantitative Structural Activity Relationship (QSAR) model generation through previously synthesized compounds. The 2-D QSAR model generated is then engaged for the predicting of the activity of our proposed compounds to be synthesized. This ligand-based approach of computer aided drug designing (CADD) is complimented further with the molecular docking simulations. Molecular docking of our proposed compounds was done to study the interaction of these compounds with the target protein i.e. tyrosine kinase receptor. Almost all the compounds showed significant results. Among them the most potent compound is SSIV which has -11.8 K/Cal/Mole.
KEYWORDS:Cancer; CADD; 2D QSAR; In Silico; Indole
Download this article as:| Copy the following to cite this article: Rana M, Ghosh N. S, Kumar D, Singh R, Monga J. 2D-QSAR Assisted Design, and Molecular Docking of Novel Indole Derivates as Anti-Cancer Agents. Orient J Chem 2024;40(5). |
| Copy the following to cite this URL: Rana M, Ghosh N. S, Kumar D, Singh R, Monga J. 2D-QSAR Assisted Design, and Molecular Docking of Novel Indole Derivates as Anti-Cancer Agents. Orient J Chem 2024;40(5). Available from: https://bit.ly/4feWWdV |
Introduction
Cell cycle and apoptosis are two utmost important processes of human cell growth and programmed cell death 1. Cancer is considered a complex disease which occurs when human body fails to regulate these two processes 2. These uncontrolled dividing cancer cells hijacks the process of normal cell division3. As per the WHO, cancer has accounted for around 10 million deaths worldwide in 2020. Most common types of cancers are rectum, lung, breast, prostate and colon tumors. It has also been projected by the WHO that by 2040, there will be around 16.3 million deaths per year worldwide due to this deadly disease 4.
This has posed an eminent threat and challenge to the medicinal chemists to develop novel molecules which can more effectively treat the occurrence of cancer. To serve this purpose, heterocyclic moieties have played an indispensible role in the development of many lifesaving drugs against several ailments 5. Indole scaffold is one is one of the promising heterocycles present in many drugs employed in the cancer treatment such as naturally occurring alkaloids vincristine and vinblastine 6.
 |
Scheme 1: Structure of Vincristine and Vinblastine |
Traditional drug discovery process is associated with high cost and consumes lots of time for the development of any novel potential drug7.To overcome this, computer aided drug designing (CADD) approaches are employed by the medicinal chemists for reducing this cost and to reduce the overall time in the entire drug discovery process8. QSAR technique is a ligand-based drug designing (LBDD) approach of CADD in which a mathematical relationship is developed for the chemical properties of already reported compounds and their biological activities. In the similar manner, molecular docking simulation is structure-based drug designing (SBDD) method of CADD which works on lock & key mechanism in which proposed ligand is placed inside the active cavity of the receptor and Gibbs free energy (Dock Score) of the system is calculated 9.
In the current study, first a 2D-QSAR model is generated against anticancer activity and this QSAR equation generated is employed for the designing of our novel indole-based drug moieties. The newly synthesised indole-based molecules were subjected to in-vitro anticancer activity. The results obtained from the anticancer activity are further validated via molecular docking simulations.
Materials and Methods
All the computational work has been performed by using Lenovo thinkpad system having intel core i5 @ 2.40 GHz, having RAM capacity of 8GB and a 512 GB hard disk. Marvin sketch of ChemAxon has been employed for all the molecular modelling work including drawing of structures, energy minimization etc. QSARINS software of the University of Insurbia is employed to develop and externally validate the generated QSAR model10. AutoDock tools and AutoDock vina of the scripps research institute is employedin the molecular docking simulations11.
QSAR Studies
Collection of the Dataset and Data Optimization
24already reported and synthesized thiosemicarbazone-indole derivatives were utilized for the creation of the 2D-QSAR model 12. Inhibitory potential of the reported compounds was in micromolar range and exhibits the minimum required concentration of the compounds to restrain 50% of the growth of PC3 cell lines (IC50). To remove the skewness in the data, IC50 values of these 24 compounds has been changed to negative logarithm values (PIC50) 13. The chemical structures of thiosemicarbazone-indole derivatives along with their inhibitory concentrations are given in the Table 1.
 |
Table 1: Structure of the 24 compounds used for the generation along with their IC50& calculated PIC50 values |
Molecular Descriptor Calculations
Molecular descriptors of the 24 derivatives were calculated using PADEL descriptor software 14. All the structures of the 24 derivatives were drawn in the mol format and then subjected to PADEL which computes a total of 1875 descriptors which includes autocorrelation, geometrical, electrostatic, topological, spatial, constitutional and thermodynamic descriptors.
Pretreatment of the data, division of dataset and generation of QSAR equation
Before the development of the QSAR model, descriptors having almost same values and descriptors which are inter correlated were removed for the development of a robust and reliable equation. For this purpose, both constant and inter correlated descriptors showing variance more than 80% were removed. A random approach is used for dividing the dataset into training and test dataset in which 70% of the compounds divided into training and remaining 30% were divided into test data set. For generating the QSAR model, a search heuristic approach called as Genetic Algorithm (GA) is used which mimics the techniques of natural selection like inheritance, crossover, mutation, and selection.
Internal Validation
The cross-validation method was used for the assessing the predictability ofcreated QSAR equation through internal validation. The following equation is used for calculating the cross validated Q2cv:
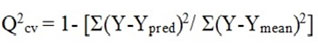
Here, Y represents the experimental biological activity value (PIC50),Ypred stands for predicted biological activity by QSAR model &Ymeanstands for the average of Y of the training set compounds.
Another parameter for assessing the quality and reliability through internal validation is squared correlation coefficient R2 value of the training set. But this value can be biased as its value is not as reliable if we increase the quantity of descriptors. To prevail over this hindrance, a freshfactor R2adj is used which is calculated as follows:
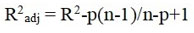
Where p is the number of the descriptors employed and n is the number of compoundsemployed in the training set for the generation of QSAR model. There is an acceptable fact that if difference between R2 and R2adj is less than 0.3 then we can infer that numbers of descriptors selected are acceptable 9.
External Validation
For assessing a QSAR model for its robustness, Golbraikh and Tropsha has given some statistical parameters 15 which are given in Table 2. Where R20 is coefficient of squared correlation among experimental and predicted values and R’20 is same among predicted and experimentalvalues of test set.
Table 2: Golbraikh and Tropsha parameters for the validation of the 2D QSAR model
|
S.No. |
Parameter |
Threshold value |
|
1 |
Q2 |
Threshold value Q2>0.5 |
|
2 |
R2train |
Threshold value R2train > 0.6 |
|
3 |
|R20 – R,20| |
Threshold value |R20 – R,20|< 0.3 |
|
4 |
K or k’ |
0.85< K<1.15 and 0.85 <k’ <1.15 |
|
5 |
R2test -R20/ R2test |
Threshold value R2-R20/ R2<0.1 |
Y randomization Test
Y randomization test is done to evaluate that QSAR equation generated is not resulted through by a fluke instead is a robust model. This test is performed by shuffling the value of biological activity while keeping the values of descriptors constant. This shuffling is done n number of times and robustness of the developed model is assessed through comparing R2 and Q2 of Y randomized equations with original QSAR equation and it should be as low as possible 16.
Applicability Domain
Applicability Domain (AD) is a chemical space of developed QSAR model where all the predictions done by the model is of the utmost accuracy. As per the 3rd principle of Organization for economic Co-operation and development (OECD), it is highly suggested to describeAD of a QSAR equation. AD is used intended for identifying response outliers as well as influencers in QSAR equation17.
In the current study, Williams plot and insurbia graph is employed for defining AD of the formed QSAR model 10. This is a simple approach in which every new chemical is defined whether it will be within the AD or will be an outlier. The leverage hi of each chemical of training as well prediction dataset is calculated as follows:
hi = xit(XtX)xi
Where xi is the descriptor vector of the under consideration data point, Xasthe descriptor matrix and Xtas the transpose of the descriptor matrix. The threshold leverage h* is calculated as
h*= 3(p+1)/n
Where, p = number of variables
n = the number of compounds in the training set
For every chemical the value of hi calculated should be less than the threshold value, otherwise it is considered as outside the AD but if it has small standardized residual than it may not be considered as outlier. For standardized residuals a cut-off value of ±3 is considered to be inside the AD.
Predicting the biological activity of the designed molecules
Biological activity of all our proposed 11 compounds was predicted from the mathematical equation obtained from our QSAR model developed. Initially, molecular descriptor calculation was performed of these derivatives using PADEL software and then substituting the values of these descriptors in the QSAR equation we obtained our predicted biological activity.
In-silico molecular docking analysis
The drawing of the molecular structure & their initial 3D optimization is performed on the marvinsketch of Chemaxon. Molecular docking of all our proposed molecules is performed against the tyrosine kinase receptor (PDB ID 6Z4B) and taking Osimertinib as the reference for comparative study. All the ligand & Protein preparation steps were performed using AutoDock tools 1.5.6 whereas Molecular docking was done by employingAutoDock vina of The Scripps Research Institute.
Results
QSAR studies
The QSAR model was generated through employing Genetic Algorithm (GA) to get the multiple linear regression (MLR)model. 3 descriptors were used for generating the QSAR equation. The equation of developed QSAR model is given as follows:
PIC50 = 16.61 – 0.8581ATSC3e -8.8485GATS8v – 0.5174nHBDon_Lipinski (Eq.1)
Where, Ntrain:17, R2: 0.8622 ,R2adj: 0.8304, Q2loo: 0.7730, Ntest: 07, R2test: 0.7770 & MAE (external): 0.2784. From looking above the QSAR equation, it is evident that the all of the descriptors employed for the generation of the model have contributed negatively in the biological activity. The details of the descriptors employed have been given in the table 3. The graph between the predicted and observed PIC50 values of the molecules employed for generation of the equation is given in the Figure 1.
 |
Figure 1: The predicted and observed PIC50 values of the compounds employed for the generation of the 2D QSAR model obtained from the QSARINS software. |
Table 3: The types of descriptorsthat were employed for the generation of the 2D QSAR model
|
S. No. |
Name of Descriptor |
Type |
Description |
Contribution |
|
1 |
ATSC3e |
2D |
Autocorrelation |
Negative |
|
2 |
GATS8v |
2D |
Geary autocorrelation of lag 8 weighted by van der waals volume |
Negative |
|
3 |
nHBDon_Lipinski |
2D |
Number of Hydrogen Bond Donors |
Negative |
The quality of any QSAR equationdeveloped is assessed both internally and externally. For the validation of the equation internally, our QSAR equation possesses R2: 0.8622 &R2adj: 0.8304 values respectively which signifies that predicted biological activity of the developed is well correlated with the experimental values. Further the robustness of the model and validation that the current model is not developed by fluke is done through Y randomization test. In this, we developed 50 random QSAR models and their values of R2& Q2 clearly suggests that they are far behind the values obtained from our original 2D-QSAR equation(Figure 02).
 |
Figure 2: Y scrambling plot of generated 2D QSAR modelobtained from the QSARINS software |
In defining the Applicability Domain (AD), none of the moleculesused for the development of QSAR model falls outside the AD. This clearly suggests that our QSAR model has a great predictability evident through the Williams plot (Figure 03).
 |
Figure 3: Williams plot for AD of the generated 2D QSAR modelobtained from the QSARINS software |
The criteria given by the Golbraikh and Tropsha for validating any QSAR model externally are the most acceptable parameters till date. Our model has clearly passes all the criteria set by theGolbraikh and Tropsha for a successful QSAR model (Table 4)
Table 4: Golbraikh and Tropsha parameters obtained of the developed QSAR model.
|
S.No. |
Parameter |
Threshold value |
Model Score |
|
1 |
Q2 |
Threshold value Q2>0.5 |
0.7730 |
|
2 |
R2test |
Threshold value R2test > 0.6 |
0.8622 |
|
3 |
|R20 – R,20| |
Threshold value |r02 – r’02|< 0.3 |
0.0318 |
|
4 |
K or k’ |
0.85< k<1.15 and 0.85 <k’ <1.15 |
0.9967 or 1.0007 |
|
5 |
R2test -R20/ R2test |
Threshold value R2-R20/ R2<0.1 |
0.04138 |
Virtual Screening &in-silico Docking Analysis
The 2D-QSAR model developed in ourresearch is further used for virtual screening through by predicting the PIC50 value of our proposed molecules. The predicted PIC50 of the proposed compounds is given in the Table 05. In the virtual screening, all our compounds have shown remarkable predicted biological activity except the compounds 1R & 1U.
Table 5: Predicted PIC50 values of synthesized molecules along with values of their descriptors.
|
S.No. |
Name |
R |
ATSC3e |
GATS8v |
nHBDon_Lipinski |
Predicted activity (PIC50) |
|
1. |
SS-1D |
H |
0.01801 |
1.18328 |
3 |
4.575756 |
|
2. |
SS-1E |
4-OH |
-0.3156 |
1.16946 |
4 |
4.466878 |
|
3. |
SS-1H |
2,6-di-hydroxy |
-0.14666 |
1.128865 |
5 |
4.163588 |
|
4. |
SS-1N |
2-ethyl |
0.232791 |
1.161088 |
3 |
4.587754 |
|
5. |
SS-1O |
4-amino |
-0.01767 |
1.124904 |
5 |
4.087935 |
|
6. |
SS-1R |
3,5-diamino |
-0.1903 |
1.212957 |
7 |
2.422408 |
|
7. |
SS-1S |
3,5-dichloro |
-0.35424 |
1.152108 |
3 |
5.170913 |
|
8. |
SS-1U |
4-amino-2-hydroxy |
-0.16527 |
1.116822 |
6 |
3.768681 |
|
9. |
SS-1V |
3-methoxy-2-nitroxy |
0.440941 |
1.111587 |
3 |
4.846998 |
|
10. |
SS-1X |
3-formyl |
0.131693 |
1.166851 |
3 |
4.62353 |
|
11. |
SS-1Y |
4-formyl-3-hydroxy |
-0.00632 |
1.132459 |
4 |
4.528773 |
The proposed compounds were further evaluated via molecular docking analysis to study their interactivity with the receptor. The results of our docking analysis are given in the Table 06.
Table 6: The dock score of the synthesized compounds along with their interactions with the different amino acids.
|
S.No. |
Compound Name |
Dock Score (KCal/Mole) |
H-Bond Number |
Amino acid Residues involved in Hydrogen Bonding |
Amino Acids involved in the interaction with ligand |
|
1. |
Osimertinib |
-9.4 |
01 |
LYS745 |
ILE759, LEU777, MET766, LEU788, LYS745, MET790, LEU718, LEU844, VAL726, ALA743, |
|
2. |
SS-1D |
-10.7 |
01 |
LYS745 |
LEU777, LEU788, MET766, LEU858, LYS745, ASP855, MET790, LEU844, VAL726, ALA743, CYS797 |
|
3. |
SS-1E |
-10.9 |
00 |
NIL |
LEU777, LEU788, MET766, PHE856, VAL726, LEU718, LEU797, ALA743, MET790, LYS745 |
|
4. |
SS-1H |
-11.2 |
01 |
LYS745 |
MET790, LEU777, LEU788, MET766, ALA743, VAL726, LEU797, CYS797, LEU858 |
|
5. |
SS-1N |
-10.6 |
00 |
NIL |
MET790, LEU777, LEU788, LYS745, ALA743, VAL726, LEU844, LEU718, GLY719, CYS797 |
|
6. |
SS-1O |
-10.9 |
00 |
NIL |
LEU777, LEU788, MET766, MET790, LYS745, ALA743, VAL726, CYS797, LEU718, LEU844 |
|
7. |
SS-1R |
-11.1 |
01 |
PHE856 |
LEU777, LEU788, MET766, MET790, LYS745, ALA743, VAL726, LEU718, LEU844 |
|
8. |
SS-1S |
-11.0 |
00 |
NIL |
MET790, LYS745, ALA743, VAL726, LEU743, MET793, LEU792, LEU718, LEU844, CYS797, ASP855, LEU788, LEU861, LEU862, MET766, LEU861 |
|
9. |
SS-1U |
-11.3 |
01 |
LYS745 |
LEU777, LEU788, MET766, MET790, ASP855, VAL726, LEU743, CYS797, LEU844 |
|
10. |
SS-1V |
-11.8 |
00 |
NIL |
LEU777, LEU788, MET766, LEU861, LEU862, LEU858, LEU743, VAL726, MET790, LYS745, CYS797, LEU844, LEU718 |
|
11. |
SS-1X |
-10.8 |
00 |
NIL |
LYS745, VAL726, MET790, LEU844, LEU718, LEU788, LEU743, LEU747, LEU861, LEU862, LEU858, MET766 |
|
12. |
SS-1Y |
-10.8 |
00 |
NIL |
LEU788, MET766, ASP855, LYS745, LEU777, LEU743, MET790, VAL726, CYS797, LEU718 |
From the docking analysis, it was interesting to see that all our proposed compounds showed higher dock score when compared to the Osimertinib the reference standard used in the molecular docking analysis. The highest docking store was shown by the compound 1V having the dock score of -11.8 Kcal/mole but this compound didn’t show any hydrogen bond interaction (Figure 04).
 |
Figure 4: Interaction of the compound SS-1H with the target proteinobtained from the Biovia Discovery studio academic visualizer |
The standard used Osimertinib has shown one hydrogen bond interaction with the receptor amino acid Lysine745 (Figure 05). The same type of hydrogen bond interactions are also possessed by the compounds 1D, 1H & 1U with the same amino acids. The compound 1R has also possessed one hydrogen bond interaction with the amino acid Phenyl Alanine 856.
 |
Figure 5: Interaction of the Osimertinib with the target protein obtained from the Biovia Discovery studio academic visualizer |
Discussion
A robust 2-D QSAR model is developed in the current study with high predictability as evident through the validation parameters both internal and external.The Y-randomization test has also further verified that our model was not developed merely coincidentally. The graph between the predicted and experimental biological activities clearly indicates that both these values have close relationship near to the straight line.
The developed 2-D QSAR model was then employed for the screening of our proposed compounds by predicting their biological activities. This is further evaluated through the molecular docking simulations to see the interactions of our compounds with the target receptor. The molecular docking studies have shown an interesting fact that all of our proposed compounds have higher dock score when compared to the standard used Osimertinib.
Conclusion
CADD approach has become the backbone of any drug discovery process for a medicinal chemist. In the current study, we incorporated both the 2D-QSAR & Molecular docking analysis of the Ligand & Structure based drug designing approaches respectively for designing the novel indole- based compounds for the anti-cancer activity. The current in-silico studies conducted has opened new horizons for us to transfer the current research further for the synthesis and in-vitro screening. Therefore from the current study it is inferred that this study should further be shifted for in-vivo and in-vitro research against cancer.
Acknowledgment
The researchers would like to thank the Department of Pharmaceutical Sciences at Adarsh Vijendra Institute of Pharmaceutical Sciences, Shobhit University, Gangoh, Saharanpur, Uttar Pradesh, for their co-operation in this study.
Conflict of Interests
The author(s) do not have any conflict of interest.
References
- Fouad, Y A.; Aanei C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7(5), 1016–1036.
- Nam, N. H.;& Parang, K. Current targets for anticancer drug discovery. Curr. Drug Targets 2003, 4(2) ,159–179.
CrossRef - Storey S. Targeting apoptosis: Selected anticancer strategies. Nat. Rev. Drug Discov. 2008, 7, 971–972.
CrossRef - Globocan (TheGlobalCancerObservatory). AllCancers; International Agency for Research on Cancer—WHO: Lyon, France, 2020; Volume419, pp.199–200. Available online:https://gco.iarc.fr/ today/home.
- Jampilek, J. Heterocycles in Medicinal Chemistry. Molecules. 2019, 24(21), 3839. doi: 10.3390/molecules24213839.
CrossRef - Sharma P, Thakur A, Goyal A, Grewal AS. Molecular docking, 2D-QSAR and ADMET studies of 4-sulfonyl-2-pyridone heterocycle as a potential glucokinase activator. Results in Chemistry. 2023 Dec 1;6:101105..
CrossRef - Yu,W.;MacKerell, A.D. Computer-aided drug design methods. Antibiotics: methods and protocols. 2017, 1520, 85-106. doi:https://doi.org/10. 1007/978-1-4939-6634-9_5
- Surabhi S.; Singh, BK. Computer aided drug design: An overview. J. drug deliv. ther. 2018: 8(5), 504–509. doi https:// doi.org/10.22270/jddt.8i5.1894
CrossRef - Thakur A.; Sharma B.; Parashar A.; Sharma V.; Kumar, A.; Mehta V. 2D-QSAR, molecular docking and MD simulation based virtual screening of the herbal molecules against Alzheimer’s disorder: an approach to predict CNS activity. J. Biomol. Struct. Dyn. 2023. DOI: 10.1080/07391102.2023.2192805
CrossRef - Gramatica P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26(5), 694–701. https://doi.org/10.1002/qsar.200610151
CrossRef - Trott, O.; Olson, A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010: 31(2) 455-461.
CrossRef - He, Z.X.; Huo, J.L; Gong, Y.P.; An, Q.; Zhang, X.; Qiao, H.; Yang, F.F.; Zhang, X.H.; Jiao, L.M.; Liu, H.M.; Ma LM, Zhao W, Design, synthesis and biological evaluation of novel thiosemicarbazone-indole derivatives targeting prostate cancer cells, EurJMed Chem. https://doi.org/10.1016/j.ejmech.2020.112970.
CrossRef - Thakur, A.; Kumar, A.; Sharma, V.K; Mehta V. PIC50: An opensource tool for interconversion of PIC50 values and IC50 for efficientdata representation and analysis. BioRxiv, 2022, 10. https://doi.org/10.1101/2022.10.15.512366
CrossRef - Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011: 32(7) 1466–1474. DOI https://doi.org/10.1002/jcc.21707
CrossRef - Golbraikh,A.;Tropsha, A. Beware of q2. J Mol Graph Model. 2002: 20(4):269-76. DOI https://doi.org/10.1016/s1093- 3263(01)00123-1
CrossRef - Rucker, C; Rucker G, Meringer M. y-Randomization and its variants in QSPR/QSAR. J Chem Inf Model 2017 47(6) 2345–2357. https://doi.org/10.1021/ci700157b.
CrossRef - Roy, K.; Kar,S;Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. https://doi.org/10.1016/j.chemolab.2015.04.013
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


