From Molecules to Medicine: The Remarkable Pharmacological Odyssey of Quinoline and Its Derivatives
Neil B. Panchal1 and Vipul M. Vaghela2
Department Of Pharmacy, Sumandeep Vidyapeeth Deemed to be University, Piparia, Waghodia, Vadodara, Gujarat, India.
Department Of Pharmaceutical Chemistry, A. R. college of pharmacy and G. H. Patel institute of pharmacy, Vallabh Vidyanagar, Anand, Gujarat, India.
Corresponding Author E-mail: neil.dop@sumandeepvidyapeethdu.edu.in
DOI : http://dx.doi.org/10.13005/ojc/390303
Article Received on : 09 Apr 2023
Article Accepted on : 15 May 2023
Article Published : 03 Jul 2023
Reviewed by: Dr. Gavat Cristian
Second Review by: Dr. Mujeeb Ahmad
Final Approval by: Dr. Dinesh Chand Sharma
Quinoline, a nitrogen-containing heterocyclic molecule, has emerged as an important scaffold in medicinal chemistry due to its diverse pharmacological effects. The fused quinazoline nucleus in particular has garnered attention for its potent properties, including antibacterial, antiviral, anti-cancer, anti-inflammatory, antioxidant, and anticonvulsant effects. The mechanism of action of these quinoline derivatives is specific to their pharmacological activity.
Researchers have studied the chemical and pharmacological properties of quinoline derivatives extensively, with a focus on their anticancer activity. Their ability to bind with DNA, impede DNA synthesis, and cause oxidative stress has made them promising candidates for cancer therapy. The quinoline nucleus's unique chemical structure and flexibility of substituents provide a vital component in drug discovery research.
This review article provides a comprehensive synthesis of the chemical and pharmacological properties of quinoline derivatives. The study highlights the immense potentiality of this ring system and their pharmacological scaffold. The review covers the diverse biological activity of quinoline derivatives and their mechanism of action, including their effects on DNA synthesis, cell division, virus replication, adhesion to host cells, cytokine generation, gene modulation, free radical scavenging, and neurotransmitter activation.
The ability to modify the quinoline nucleus with different substituents around the centroid has made it a privileged scaffold for researchers to work with. Researchers have created novel therapeutic compounds with improved pharmacological characteristics, leading to innovative therapies for various disorders. Further research into synthesis, reactions, and pharmacological effects of quinoline derivatives can pave the way for the development of new drugs.
The article specifically focuses on the anticancer activity of quinoline derivatives, highlighting their potential as a viable clinical candidate for cancer therapy. The review emphasizes the importance of the quinoline nucleus as a key scaffold for drug discovery research and encourages further exploration of its chemical and pharmacological properties. In conclusion, this review article provides a comprehensive overview of the immense potential of quinoline derivatives and their significance in medicinal chemistry research.
Anti-cancer; DNA-interaction; Pharmacology; Quinoline derivatives; Synthesis
Download this article as:| Copy the following to cite this article: Panchal N. B, Vaghela V. M. From Molecules to Medicine: The Remarkable Pharmacological Odyssey of Quinoline and its Derivatives. Orient J Chem 2023;39(3). |
| Copy the following to cite this URL: Panchal N. B, Vaghela V. M. From Molecules to Medicine: The Remarkable Pharmacological Odyssey of Quinoline and its Derivatives. Orient J Chem 2023;39(3). Available from: https://bit.ly/3NCavHD |
Introduction
Quinoline, a heterocyclic system composed of two fused six-membered aromatic rings (benzene and pyridine), has received considerable attention in medicinal chemistry due to its diverse and distinct biopharmaceutical activities. Quinoline derivatives belong to the nitrogen-containing heterocyclic compounds and have been extensively studied for their broad range of pharmacological responses, such as anti-cancer, anti-malarial, antibacterial, antifungal-antiprozoal, anthelmintic, local anesthetic, antiasthmatic, antipsychotic, antiglaucoma, and cardiotonic activities. The presence of quinoline alkaloids in various plant species has further amplified their significance in medicinal chemistry.
The unique chemical structure of quinoline and the ability to modify its substituents have enabled researchers to design novel and potent quinoline-based drugs. The versatility of quinoline in exhibiting a multitude of pharmacological responses has captured the attention of medicinal chemists to explore its vast potential in various therapeutic applications. Ongoing investigations into the pharmacological effects of quinoline derivatives will lead to the discovery of innovative and effective therapies for various diseases.
In medicinal chemistry, understanding the structure-activity relationship (SAR) is critical for designing and developing new drugs with improved pharmacological activity. SAR studies involve evaluating the relationship between the chemical structure of a molecule and its pharmacological activity. Researchers have conducted numerous SAR studies of quinoline derivatives to identify the specific structural features that are responsible for their diverse pharmacological effects.
For instance, SAR studies of quinoline-based antimalarial drugs have highlighted the importance of a basic nitrogen atom in the quinoline ring for antimalarial activity. The presence of a halogen atom in the molecule can also significantly enhance its activity against malaria. Similarly, in the case of quinoline-based anticancer drugs, SAR studies have demonstrated that the presence of a hydroxyl or methoxy group at position 7 on the quinoline ring can improve the compound’s antitumor activity. The introduction of a substituent at position 4 on the quinoline ring can also enhance the compound’s potency against cancer cells.
In the case of quinoline-based antibacterial drugs, SAR studies have identified the importance of the substitution pattern around the quinoline ring for antibacterial activity. The introduction of a fluorine atom at position 6 on the quinoline ring can significantly enhance the compound’s antibacterial activity.
Overall, SAR studies have provided valuable insights into the molecular mechanisms underlying the pharmacological activity of quinoline derivatives. By identifying the specific structural features responsible for a compound’s pharmacological activity, researchers can design and develop new drugs with improved efficacy and reduced side effects1
Quinoline
 |
Figure 1: Quinoline moiety |
Quinoline is an exceptional organic compound that belongs to the class of nitrogen-containing heterocyclic substances. Its distinct molecular structure is characterized by the fusion of two aromatic rings, namely a benzene ring and a pyridine ring. This bicyclic compound is also referred to as benzo[b]pyridine and is considered as an analogue of naphthalene (1-azanapthalene). Its molecular weight is 129.16, and it has a log P value of 2.04, which indicates its lipophilicity. Quinoline is a weak tertiary base.31 Due to its unique structure, quinoline exhibits both electrophilic and nucleophilic substitution reactions, allowing for various modifications of its substituent groups. Quinoline exhibits a remarkable ability to form salts with acids and display analogous chemical reactions to those observed in pyridine and benzene, showcasing its versatility as a heterocyclic compound.2
Physical properties3,4
A distinctive odor accompanies the colorless, hygroscopic liquid, which turns brown when exposed to light. Its solubility in both water and a wide range of organic solvents highlights its versatility as a chemical compound.
More sol in hot than cold water; sol in ethanol, ethyl ether, acetone, carbon disulfide and other common organic solvents.
Boliling point: 237.1 °C
Melting point: -15 °C
Relative density: Relative density (water = 1): 1.09g/ml
Synthesis
 |
Figure 2: Quinoline synthesis. |
Skarup Synthesis
is a well-known method for the synthesis of quinoline and its derivatives. The reaction proceeds by treating aniline or its derivatives with a mixture of glycerol, concentrated sulfuric acid, and nitrobenzene in the presence of ferrous sulfate. The reaction is believed to proceed through several intermediate steps, including the formation of a nitroso compound, which undergoes cyclization to form the quinoline ring system.It is important to note that this reaction requires careful handling of the reactants, as concentrated sulfuric acid is a strong acid and can be dangerous if not used properly.5,6 Additionally, the reaction conditions may need to be optimized for specific substrates to achieve high yields and purity of the desired products.In the case of o-aminophenol, the reaction proceeds through an intramolecular cyclization mechanism, in which the hydroxyl group on the phenol ring reacts with the amino group to form the quinoline ring system. The resulting product is 8-hydroxyquinoline, which has a hydroxyl group attached to the eighth carbon atom of the quinoline ring2.
 |
Figure 3: Skarup Sythesis |
Mechanism
Step 1: Glycerol (Propane-1,2,3-triol) undergoes dehydration with sulphuric acid to give acryladehyde.
 |
Scheme 1 Click here to View Scheme |
Step 2: Aniline adds to Acrolein by 1,4 –Addition.
 |
Scheme 2 Click here to View Scheme |
Step 3: Undergoes ring Closures
 |
Scheme 3 Click here to View Scheme |
Step 4: Oxidation of 1,2-dihydroquinoline
 |
Scheme 4 Click here to View Scheme |
Knorr Quinoline Synthesis
The Knorr Quinoline Synthesis is a widely recognized method for synthesizing quinoline derivatives that have diverse applications in medicinal chemistry and materials science. The reaction involves the condensation of an aromatic amine with a β-ketoester, which undergoes ring closure to form the quinoline ring system. A mild acid catalyst, such as sulfuric acid, is typically used to facilitate the formation of the imine intermediate and promote the ring closure reaction.The versatility of this synthetic method lies in the ability to optimize the reaction conditions for specific substrates, leading to high yields and purity of the desired products. By using different aromatic amines and β-ketoesters, various substituted quinoline derivatives can also be produced. However, the synthesis of the β-ketoester substrate can be challenging, which is a limitation of this method. The reaction can also be sensitive to changes in pH and temperature, affecting yield and selectivity.7,8
Despite its limitations, the Knorr Quinoline Synthesis remains a pivotal synthetic pathway for the production of quinoline derivatives. The method’s versatility, coupled with its wide-ranging applications in crucial fields, underscores the importance of this synthetic method in advancing scientific research and development.3,7,8.
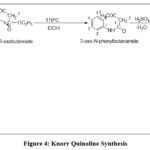 |
Figure 4: Knorr Quinoline Synthesis |
Conard –Limpach Synthesis
The Conard-Limpach synthesis is an established method for synthesizing quinoline derivatives that has found extensive use in organic chemistry. The reaction involves the condensation of aniline with a β-ketoester, which undergoes ring closure at high temperatures to form the quinoline ring system. A strong acid catalyst, such as hydrochloric acid or sulfuric acid, is typically used to promote the formation of the imine intermediate and facilitate the ring closure reaction.A unique feature of the Conard-Limpach synthesis is its ability to produce a wide range of substituted quinoline derivatives. By using different aromatic amines and β-ketoesters, the reaction can be modified to yield various substituted quinoline derivatives with different functional groups and substitution patterns.9–11 Mechanistic studies have been carried out to understand the reaction mechanism and identify key intermediates.Despite its usefulness, the Conard-Limpach synthesis has some limitations. The reaction requires high temperatures, which may restrict its application to sensitive substrates. Additionally, the reaction’s selectivity and yield may be affected by factors such as pH and the nature of the catalyst used. Nonetheless, the method’s versatility in producing substituted quinoline derivatives and its role in advancing scientific research make it an important synthetic pathway in organic chemistry3,10,11.
 |
Figure 5: Conard –Limpach Synthesis |
From Indole and dichloromethylene in the presence of methyl lithium3.
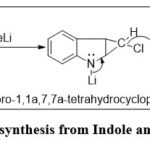 |
Figure 6: Quinoline synthesis from Indole and dichloromethylene |
The Friedlander synthesis
The Friedländer synthesis is a well-established method for synthesizing quinoline and its derivatives. This reaction involves the condensation of an aliphatic carbonyl compound and an aromatic carbonyl compound with an –NH2 group at the ortho position in the presence of a base. The mechanism of this reaction involves the formation of an enamine intermediate, which undergoes intramolecular cyclization to form the quinoline ring system. A strong base like potassium hydroxide or sodium hydroxide is typically used to promote the formation of the enamine intermediate and facilitate the ring closure reaction. However, the yield and selectivity of the reaction can be affected by the choice of base and reaction conditions. The Friedländer synthesis can synthesize a broad range of quinoline derivatives, including various substituted quinolines, isoquinolines, and quinazolines. The reaction can be tailored by using different aliphatic and aromatic carbonyl compounds to produce substituted quinoline derivatives with diverse functional groups and substitution patterns. Mechanistic studies have proposed that the reaction proceeds through a stepwise mechanism. The Friedländer synthesis has some limitations such as the need for high reaction temperatures and long reaction times, which restricts its utility for sensitive substrates. Additionally, the reaction can be sensitive to reaction conditions like pH and the nature of the base used, which can impact the yield and selectivity of the reaction.3.
 |
Scheme 5 |
Reaction Mechanism
 |
Figure 7: The Friedlander synthesis |
Chemical properties2:The most typical reaction for quinoline are:
- Heteroatom reactions
- Electrophilic and nucleophilic substitution reactions
- Oxidation and reduction
Heteroatom reactions
Quinoline is a versatile compound with a nitrogen atom in its ring structure that can undergo different reactions, including protonation, alkylation, and acylation, to create a diverse range of quinoline derivatives. The protonation of quinoline can form quinolinium cations, which can undergo nucleophilic substitution or oxidative addition reactions to generate various derivatives. Alkylation and acylation reactions can also produce quinoline derivatives with different functional groups that can be useful in synthesizing organic compounds.However, the lower basicity of quinoline compared to pyridine can affect its reactivity in reactions like nucleophilic substitution and addition. Quinoline’s weaker ability to donate electrons to proton acceptors can also affect the selectivity and rate of alkylation or acylation reactions. Nonetheless, quinoline and its derivatives have shown promise in the development of novel anticancer compounds to combat drug resistance and improve healthcare expenditure. With further research, the potential applications of quinoline derivatives in medicine and other fields can be explored2.
 |
Figure 8: Heteroatom reactions of Quinoline |
Electrophilic substitution reactions
Quinoline undergoes substitution reactions in the benzene ring preferably at positions 8 and 52.
 |
Figure 9: Electrophilic substitution reaction of Quinoline |
Nucleophilic substitution reaction
Quinoline readily gives nucleophilic substitution reaction at 2nd position. If this position is blocked the reaction occurs at 4th position2.
 |
Figure 10: Nucleophilic substitution reaction of Quinoline |
Oxidation
Quinoline is a nitrogen-containing heterocycle with a pyridine ring that is relatively resistant to oxidation due to the delocalization of the lone pair of electrons on the nitrogen atom into the aromatic ring system. This electron delocalization stabilizes the pyridine ring and makes it less available for nucleophilic attack, which results in quinoline being a weaker base than other nitrogen-containing heterocycles. The benzene ring in quinoline can be oxidized in the presence of strong oxidizing agents such as potassium permanganate, chromic acid, or nitric acid. This oxidation leads to the formation of a diol intermediate that undergoes cleavage to yield a carboxylic acid and an aldehyde. This reaction provides a useful method for the synthesis of carboxylic acids and aldehydes from quinoline derivatives. Despite the resistance of the pyridine ring to oxidation, the benzene ring in quinoline remains susceptible to oxidative attack. This susceptibility can be attributed to the relative instability of the benzene ring compared to the pyridine ring. Thus, the reactivity of the benzene ring in quinoline can be exploited in the synthesis of carboxylic acids and aldehydes. In summary, the resistance of quinoline to oxidation is due to the stabilization of the pyridine ring by the delocalization of the lone pair of electrons on the nitrogen atom into the aromatic ring system. This electron delocalization makes quinoline a weaker base than other nitrogen-containing heterocycles. However, the benzene ring in quinoline remains susceptible to oxidation and can be oxidized to form useful intermediates. 2
 |
Figure 11: Oxidation reaction of Quinoline |
Reduction
Quinoline can be partially or fully reduced under different conditions, but the reduction of the pyridine ring is more difficult than the benzene ring due to nitrogen atom deactivation. Partial reduction can be achieved with mild reducing agents, while full reduction requires more rigorous conditions like metal hydrides. Partially reduced quinolines, such as dihydroquinolines and tetrahydroquinolines, are useful building blocks for natural products synthesis, while fully reduced quinolines like piperidine derivatives can be found in pharmaceuticals and bioactive compounds. However, the reduction of quinoline can be challenging due to over-reduction or incomplete reduction, so selecting the right reducing agent and reaction conditions is essential to achieving the desired degree of reduction and minimizing side reactions. The synthetic utility of quinoline reduction makes it an important tool in the preparation of diverse structures. 2
 |
Figure 12: Reduction reaction of Quinoline |
Pharmacological scaffold
Extensive research in the last decade has focused on the pharmacological potential of quinoline’s scaffold, leading to the discovery of its ability to produce and isolate various pharmacologically active compounds. Quinoline derivatives have shown diverse biological activities, including antibacterial, antimalarial, anticancer, anti-inflammatory, and antiviral effects. This has increased interest in quinoline as a scaffold for drug discovery due to its unique structural features, allowing for the design and synthesis of novel bioactive compounds. Numerous quinoline-based drugs have been developed, demonstrating promising results in preclinical and clinical studies, highlighting the importance of this scaffold in drug discovery research.1,12–14
Modifications to the quinoline scaffold have been extensively studied to improve its pharmacological properties, including solubility, bioavailability, and selectivity. Computational methods have facilitated the design of novel quinoline derivatives with enhanced pharmacokinetic and pharmacodynamic properties. The study of quinoline and its derivatives has provided valuable insights into the development of new drugs with diverse pharmacological effects, revealing new therapeutic targets for a wide range of medical conditions. Further research in this area is expected to lead to the discovery of new therapeutics and the advancement of modern medicine.15,16
 |
Figure 13: Pharmacologcal scaffold |
Anticancer Activity
Despite advancements in diagnosis and treatment, cancer remains a major public health concern that poses significant challenges for individuals and society as a whole. One key class of chemotherapeutic agents used in cancer treatment is cytotoxic drugs, which target the DNA replication process to inhibit cancer cell proliferation.17–19
Cancer cells require a constant supply of new DNA to support their rapid growth, and cytotoxic drugs damage the DNA to trigger the cell’s natural repair mechanisms. If the damage is too severe, the cell will ultimately die. However, these drugs can also harm healthy cells, leading to various side effects that impact the patient’s quality of life. Moreover, drug resistance can develop in cancer cells, reducing the efficacy of these drugs over time.19,20
Nonetheless, cytotoxic drugs remain essential in the fight against cancer, and researchers are working to develop novel drugs that are more effective, better tolerated, and less prone to resistance. Antimetabolites, alkylating agents, and DNA-binding agents are among the different types of molecules that can interfere with DNA replication, with each having its advantages and disadvantages. The decision to use a particular type of molecule depends on factors such as the type and stage of cancer being treated and the patient’s overall health. These molecules are often used in combination with other drugs or therapies to maximize effectiveness and minimize side effects.21–23
Quinoline, a versatile scaffold, shows promise in the development of new anticancer drugs. Since cancer accounts for a substantial portion of global mortality, researchers are turning to novel anticancer compounds, including quinoline derivatives, to combat multidrug resistance and optimize healthcare expenditure.24–28
Structure activity relationship
The presence of a quinoline ring with various functional groups and substitutions greatly influences the anticancer activity of the compound. For example, the compound 5-(3,4,5-trimethoxyphenyl)-8-hydroxyquinoline has been found to exhibit potent anticancer activity due to the presence of a hydroxy group in position 8 and a trimethoxyphenyl group in position 5 of the quinoline ring12,14.
The introduction of a hydrophobic group such as an alkyl chain in the quinoline ring also enhances its anticancer activity by improving its binding affinity to the target receptor. For example, the compound 5-(quinolin-8-yloxy)pentanoic acid has been found to exhibit potent anticancer activity due to the presence of a pentanoic acid chain in the quinoline ring.12,15
Quinoline derivatives with a planar structure and an extended conjugation system exhibit better anticancer activity due to their ability to intercalate with DNA. For example, the compound 2,4-dimethoxy-8-(4-methoxyphenyl)quinoline has been found to exhibit potent anticancer activity due to the presence of a planar structure and an extended conjugation system.14,29
The position of the substituents on the quinoline ring also affects the anticancer activity. For example, compounds with substitutions in positions 2 and 3 have been found to be more active against certain cancer cell lines than those with substitutions in positions 4 and 8.13,30
The presence of a halogen group in the side chain of the quinoline derivative has been found to improve its anticancer activity by increasing its lipophilicity and cellular uptake.31,32
The stereochemistry of the quinoline derivative also plays a role in its anticancer activity. For example, compounds with an (R)-configuration at the chiral center of the side chain have been found to be more active than those with an (S)-configuration1,33
The presence of a bulky group such as a tert-butyl group in the side chain of the quinoline derivative has been found to decrease its activity by decreasing its flexibility and accessibility to the target receptor.34
Literature Review
Assefa H et al. conducted a study where they performed 3D-QSAR and synthesized 4-anilinoquinazoline and 4-anilinoquinoline as an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors. They identified a number of potential derivatives that showed promise in inhibiting the EGFR tyrosine kinase enzyme. These derivatives have been reported to have an exceptional potential and having potent inhibitory activity against EGFR tyrosine kinase. The study provides valuable insight into the development of novel and effective inhibitors for the treatment of various types of cancer, particularly those that are associated with overexpression of EGFR35.
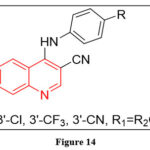 |
Figure 14 |
Chen et al. conducted a study in 2004 on the synthesis of several 11-substituted 6H-indolo[2,3-b]quinolones and prepared their methylated derivatives too. The researchers observed that the 5-methylated derivatives 14a, 15a, 16a were more cytotoxic than their respective 6-methylated counterparts 14b, 15b, 16b, and 6H-indolo[2,3-b]quinoline precursors. Among the synthesized derivatives, 11-(4-methoxyanilino)-6-methyl-6H-indolo[2,3-b]quinoline (16a) exhibited the most cytotoxicity with a mean GI50 value of 0.78µM. Moreover, it showed selective cytotoxicity for HL-60(TB), K-562, MOLT-4, RPMI-8226, and SR with GI50 values of 0.11, 0.42, 0.09, 0.14, and 0.19µM, respectively. The results suggest the potential of 11-substituted 6H-indolo[2,3-b]quinolones and their methylated derivatives, especially 5-methylated derivatives, as anti-cancer agents, with 11-(4-methoxyanilino)-6-methyl-6H-indolo[2,3-b]quinoline (16a) showing promising cytotoxicity against several cancer cell lines36.
 |
Figure 15 |
Chi et al. (2012) reported the synthesis of a set of substituted quinolines using a scalable sequence of reactions starting from 4-acetamidoanisole. and screened them for anti-breast cancer activity using T-47D breast cancer cells. The results indicated that from the tested quinolines, compound 6, 4-methyl-6-methoxy-8-[(furan-2-ylmethyl)amino]-5-(3-trifluoromethylphenoxy)quinoline, exhibited the lowest IC50 value of 16 ± 3 nM, indicating its potent anti-breast cancer activity. The results of this study point out that substituted quinolines hold potential as effective agents in the treatment of breast cancer therapy 37.
Compound 6
 |
Figure 16 |
Ghorab et al. (2007) synthesized a series of newly developed derivatives that contained N-(quinolin-1-yl)-4-toluenesulfonamide. and 4-methylbenzenesulfonamide-N-(7H-pyrimido[4,5-b]quinolin-10-yl). A set of moieties were synthesized and evaluated for their anticancer potential against Ehrlich ascites carcinoma cells. Compounds 11a, 11b, and 11c showed significant cytotoxic activity compared to Doxorubicin, indicating their potential as novel anticancer agents, necessitating further research and development.38.
 |
Figure 17 |
Scott et al. (2009) Disclosed the development of N-(4-anilinoquinolin-6-yl)acetamide derivatives as potent and selective inhibitors of CSF-1R kinase. The synthesis involved a multi-step process starting from 4-chloroquinoline. The derivatives were tested for their ability to inhibit CSF-1R kinase activity and assayed for their antitumor activity using various cell lines. Some derivatives showed excellent antitumor activity by inhibiting CSF-1R kinase. Notably, compounds 7g, 7j, and 7k exhibited potent antitumor activity against the MDA-MB-231 breast cancer cell line, with IC50 values of 18, 22, and 11 nM, respectively. Compound 7k also demonstrated significant antitumor activity against the HT-29 colon cancer cell line, with an IC50 value of 32 nM. These results suggest that amido-4-anilinoquinolines derivatives hold promise as a potential class of compounds for the development of new cancer therapies targeting CSF-1R kinase39.
 |
Figure 18 |
synthesized a series of potent inhibitors of p300/CBP histone acetyltransferases, referred to as MC compounds. These inhibitors were 4-hydroxyquinolines derivatives with a C1 to C15 alkyl chain at the C2 position and bulky functional groups at the C3 position. The authors evaluated the enzyme inhibition of these compounds using in vitro HAT assays and western blots analysis, finding that some of the compounds displayed selective inhibitory activity against the p300/CBP HAT enzymes. This study provided valuable insights into the design of inhibitors targeting histone acetyltransferases for cancer treatment.
The study investigated the potential of 4-hydroxyquinolines as inhibitors of p300/CBP histone acetyltransferases and their pro-apoptotic cytodifferentiation properties. One derivative showed promising properties, indicating its potential as a cancer treatment. The findings highlight the importance of selective enzyme inhibition in cancer therapy40.
 |
Figure 19 Click here to View Figure |
Miller et al. (2009) mentioned a novel synthesized series of 3-cyanoquinoline compounds to selectively inhibit insulin-like growth factor-1 receptor (IGF-1R), a critical mediator of cell proliferation and survival in cancer. The basic moiety is a 3-cyanoquinoline scaffold modified with various substituents. Several of the compounds showed potent inhibition of IGF-1R in vitro and in cancer cells, with the most potent having an IC50 of 0.04 nM. SAR analysis revealed that a 6-phenyl and 7-methyl substituent were important for potent activity41.
 |
Figure 20 |
Chen et al. (2009) synthesized and evaluated a series of quinoline carboxylic acid derivatives as inhibitors of insulin-like growth factors (IGFs) involved in regulating cell growth, differentiation, and survival. They designed and synthesized the compounds by modifying the quinoline scaffold at positions 2 and 3 with various substituents such as carboxylic acid, amine, and hydroxyl groups. In vitro kinase assays and cell-based assays were used to evaluate the inhibitory activity of the compounds against IGF-1R and IGF-2R. The results showed that several compounds were potent inhibitors of these receptors, with low micromolar IC50 values. The most potent compound had an IC50 of 0.17 μM and inhibited IGF-1R and IGF-2R signaling in breast cancer cells. A structure-activity relationship (SAR) analysis showed that a carboxylic acid group at position 3 of the quinoline scaffold was crucial for inhibitory activity. These findings suggest that these quinoline carboxylic acid derivatives have potential as inhibitors of IGF receptors for cancer therapy.42
 |
Figure 21 |
Ghorab et al. (2010) reported and evaluated 9H-pyrimido[4,5-b]quinoline derivatives with a sulfonamide group as potential anticancer agents against human breast cancer cells (MCF7). Compound 6 and 18 displayed similar IC50 values to the reference drug doxorubicin, while compound 8 demonstrated better activity. Moreover, compounds 8 and 18 were also assessed for their radiosensitizing effects, and both compounds showed a significant increase in radiosensitivity and enhanced cell death when combined with γ-radiation compared to radiation alone. These findings suggest that these derivatives have the potential to be developed as novel anticancer agents with radiosensitizing properties43.
 |
Figure 22 Click here to View Figure |
synthesized a series of 3,5-disubstituted and 3,5,7-trisubstituted quinolines as inhibitors of c-Met kinase, which is frequently overexpressed in human cancers. The researchers modified the quinoline scaffold with various substituents to optimize the potency and selectivity of the compounds against c-Met kinase. Through in vitro kinase assays and cell-based assays in c-Met-dependent cell lines, the synthesized compounds exhibited potent inhibition of c-Met kinase with IC50 values below 1 nM. Compound 21b showed high potency and selectivity against c-Met kinase compared to other tyrosine kinases, and inhibited c-Met phosphorylation and downstream signaling in c-Met-dependent cell lines. Additionally, the researchers evaluated the PK profile of compound 21b and found it to be favorable. This study provides significant insights into the structure-activity relationship of quinoline-based inhibitors of c-Met kinase and highlights compound 21b as a promising candidate for further development as a targeted therapy for cancer44.
 |
Figure 23 |
Patel et al. (2011) synthesized and characterized a series of s-triazine analogs and evaluated their anticancer activity against the human prostate cancer cell line DU-145 in vitro. Two compounds, 5n and 5s, exhibited high potency against DU-145 cells, with IC50 values of 0.01 and 0.02 µM, respectively, surpassing the activity of doxorubicin (IC50=0.07 µM). The compounds induced apoptosis by activating caspase-3 and caspase-9 and causing a decrease in mitochondrial membrane potential and an increase in ROS generation. This study highlights the potential of s-triazine analogs as anticancer agents for prostate cancer and provides insights into their SAR and mechanism of action45.
 |
Figure 24 |
Tzeng et al. (2012) tested the anti-proliferative activity of 6-substituted 2-(9-methoxy-11-oxoindeno[1,2-c]quinolin-6(2H)-yl)acetic acid. and found that compound 5a and 5b showed complete inhibition of cell growth at 100 µM concentration. Compound 5b showed strong anti-cancer properties by intercalating DNA, leading to inhibition of replication and transcription. It induced cell cycle arrest at G2/M phase via the ATM/Chk2 pathway, resulting in an accumulation of cells in that phase. These findings suggest the potential of 6-substituted 2-(9-methoxy-11-oxoindeno[1,2-c]quinolin-6(2H)-yl)acetic acid.as anti-cancer agents.
Compound 5b induces DNA damage, cell cycle arrest, PARP cleavage, and apoptosis through caspase activation, resulting in cell death. It shows significant anti-cancer properties and holds promise as a therapeutic agent, altough further research is needed to fully understand its efficacy and safety in vivo46.
 |
Figure 25 |
Ai Y et al. (2012) reported a group of derivatives of 9H-pyrimido[5,4-c]quinolin-4-one and evaluated their antiproliferative activity on six human cancer cell lines. The derivatives were developed with various substitutions at positions 2 and 3 to enhance their potency and selectivity. Compound 7e exhibited the most promising anti-tumor activity with broad-spectrum activity against various cancer cell lines including nasopharyngeal carcinoma, gastric cancer, lung cancer, and breast cancer. Remarkably, compound 7e displayed superior activity compared to other derivatives, indicating the importance of the specific substitutions in the compound for its potent antitumor effects. Although the study did not explore the mechanism of action of compound 7e, its substantial anti-tumor activity and broad-spectrum suggest its potential as a promising candidate for further development as a cancer therapy 47.
 |
Figure 26 |
Marganakop et al. (2012) prepared and synthesized a series of compounds with 2-chloro-3-formylquinoline thiosemicarbazones with substitutions at positions 6, 7, and 8 and screened them for anticancer activity. The study identified compounds 5a and 5b as potent anticancer agents with better drug scores and suitable c log P values. Compound 5a, with a 2-phenyl thiosemicarbazone substituent at position 8 and a methyl group at position 7, demonstrated promising anticancer activity. Similarly, compound 5b, with a 2-thienyl thiosemicarbazone substituent at position 8 and a methyl group at position 7, also showed potential as an anticancer agent. The results suggest that these compounds have the potential to be developed as lead molecules for future drug development. Overall, the study highlights the promise of 2-chloro-3-formylquinoline thiosemicarbazones with substitutions at positions 6, 7, and 8 derivatives as potent anticancer agents..48
 |
Figure 27 Click here to View Figure |
Kouznetsov et alinvestigated the anticancer activity of quinoline derivatives with substitutions at the 2nd position. The 2-α-furyl and 2-(pyridin-2-yl)quinolinederivatives, such as Compound 1, Compound 13, and Compound 14, showed significant anticancer activity against human cancer cell lines MCF-7, H-460, and SF-268, while having no cytotoxic effects on normal cells. The presence of specific functional groups in the C-2 position was found to be associated with the anticancer activity of the quinoline derivatives, and computer simulations revealed that the electronic and steric characteristics of the substituent groups could significantly impact the compounds’ anticancer activity. These findings suggest that C-2-substituted quinolines, especially 2-α-furyl and 2-(pyridin-2-yl)quinolinederivatives, have potential as selective anticancer agents.49
 |
Figure 28 |
Salih et al. (2013) discovered a novel series of quinoline and 2,3,4,5-tetrahydroquinoline derivatives with remarkable anticancer activity against HeLa, HT29, and C6 tumor cell lines. Compound-2 and Compound-6, with 6,8-dibromo-1,2,3,4-tetrahydroquinoline and 6,8-dimethoxyquinoline structures respectively, displayed potent anti-tumor effects, making them potential candidates for further investigation in cancer therapy. These findings suggest that quinoline and 2,3,4,5-tetrahydroquinoline derivatives hold great promise as effective anticancer agents that target multiple tumor cell lines, and Compound-2 and Compound-6 have the potential to be further optimized for cancer therapy.50
 |
Figure 29 Click here to View Figure |
synthesized and evaluated the anticancer efficacy of a range of trifluoromethyl quinoline derivatives containing benzenesulfonamide moiety, urea derivatives, 4-isothiocyanate, and related carbamimidothioic acid derivatives. Compound 9a exhibited higher anticancer efficacy against various cancer cell lines compared to the reference drug, doxorubicin, making it a potential candidate for further investigation in cancer treatment research. Molecular docking studies showed that these compounds might function as PI3K inhibitors, leading to cell cycle arrest and death. These findings shed light on the potential mechanism of action for the cytotoxic activity of these compounds and suggest that they may have the potential to be developed as effective anticancer agents51
 |
Figure 30 Click here to View Figure |
evaluated the potential of quinoline derivatives as telomerase inhibitors and anticancer agents in HepG2, SGC-7901, and MCF-7 cell lines. Molecules 4d and 4i showed significant anticancer activity via telomerase inhibition according to TRAP-PCR-ELISA assays. Docking simulations suggested that these compounds could bind to the telomerase active site, highlighting their potential as promising anticancer agents. The study highlights the possibility of using quinoline derivatives as telomerase inhibitors and suggests that molecules 4d and 4i have potential as new telomerase inhibitors and cancer treatments, offering great opportunities for future research in this field52.
 |
Figure 31 |
Roslonek K.B et al. (2013) implemented a study to develop new derivatives of 6H-indolo[2,3-b]quinoline with substitutions at the C-2, C-9, or N-6 positions, linked to O-L-daunosamine or L-acosamine via an alkoxy or alkyl linker. These compounds were evaluated for their cytotoxic activity against different cancer cell lines, including lung adenocarcinoma, breast cancer, melanoma, promyelocytic leukaemia, uterine sarcoma, and colon cancer, in an effort to overcome multidrug resistance. Compounds 9, 10, 12, and 17 showed promising anti-proliferative potential against all tested cancer cell lines and induced cell cycle arrest in Jurkat T-cell leukemia. This study’s novelty lies in the development of novel compounds with alkoxy or alkyl linkers and substitutions at different positions on the chromophore. This approach provides a foundation for future research to create novel molecules with enhanced anticancer potential. The study also highlights the importance of inducing cell cycle arrest as a potential mechanism of action for cytotoxic compounds, providing potential targets for future drug development53.
 |
Figure 32 Click here to View Figure |
synthesized a novel series of pyrazolo[1,5-a]quinoline pyridine derivativesand evaluated their anticancer potential against various cell lines and inhibitory effects against FabH and EGFR enzymes. The compounds demonstrated potent anticancer activity, with Compound 7k showing the strongest inhibitory activity against EGFR and Compound 7b exhibiting the strongest inhibitory activity against FabH. Molecular hybridization led to the creation of unique structures with potential therapeutic benefits, highlighting the potential of these compounds as effective anticancer agents54.
 |
Figure 33 Click here to View Figure |
Vyas VK et al. (2014) generated and commented on a series of novel 2-Quinolinecarboxamide derivatives as inhibitors of human dihydroorotate dehydrogenase (hDHODH) and anticancerous drugs. Compounds 11 and 23, which also included either a -OCH3 or -OCF3 group at the C6 position (R) and electron-withdrawing -F groups in the diphenyl ring system, were the most efficient in both screening methods. Compounds 21 and 24 also demonstrated comparable antitumor activities. This work demonstrates the compounds’ potential as hDHODH inhibitors and anticancer medicines, with ramifications for the advancement of novel cancer therapies55.
 |
Figure 34 Click here to View Figure |
Makawana A et al. (2014) investigated and effectively synthesized unique Schiff’s base analogues and assessed them for anti-cancer potential and EGFR inhibition. The preponderance of the drugs had substantial antiproliferative activity and inhibited EGFR and HER-2 activities. 5h is the most effective inhibitor among compounds tested, with IC50 value of 0.12±0.05 µM. It binds to EGFR receptor’s active pocket with low binding energy (ΔGb = -58.3691 kcal/mol) to function.. Two hydrogen bonds, dipolar pi-pi interaction, and one charge-transfer interaction. were employed in order to generate stable connections with EGFR.54
 |
Figure 35 |
Ahsan MJ et al. (2016) researched and conducted a study to evaluate the growth-inhibitory activity of two sets of heterocycles against different cell lines. They calculated LC50, TGI, and GI50 values for 10 compounds from the first set. Compound 5j was discovered to have considerable antiproliferative action, with a GI50 value of 35.1 M against HeLa cells and 60.4 M against MDA-MB-435 cells. In light of these results, it is claimed that ,5j shows promise for treating cervical cancer and melanoma 56.
 |
Figure 36 |
Shaikh S et al.(2017 synthesized a series of Tetrazolylmethyl quinolines derivatives, including compounds 6h and 6i. These compounds were found to exhibit the intriguing property of acting as covalent cross-linkers on the DNA helix, as well as intercalating within the DNA structure. Remarkably, compound 6i demonstrated a higher C score value for intercalation than 6h.
1-(1H-tetrazol-5-yl)methane. quinolines’ anticancer potential was examined by a high-dose assay on NCI-60 tumor cell lines. 6h and 6i showed remarkable growth inhibition percentages against Melanoma (SK-MEL-5) and Breast Cancer (T-47D), respectively. These derivatives could be developed into novel and effective anticancer agents.57.
 |
Figure 37 Click here to View Figure |
George RF et al. (2019) investigated the potential of quinoline-based 1H-pyrazolo[4,3-c]pyridine and their 1,3-thiazole hybrids as epidermal growth factor receptor (EGFR) inhibitors with anti-proliferative activity. The compounds were tested against various cancer cell lines and normal fibroblast WI-38 to evaluate their selectivity and safety. The study found that the compounds showed significant anti-tumor effects while remaining safe for normal cell lines. Compounds 6b, 2, and 7c were found to have potent inhibitory effects. This study suggests that quinoline-based 1H-pyrazolo[4,3-c]pyridine and their corresponding thiophene hybrids could be a promising avenue for the development of safe and effective EGFR inhibitors for cancer therapy58.
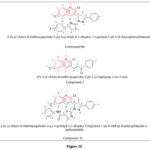 |
Figure 38 |
Othman D. et al.(2019 investigated Substituted Quinoline-thiophene scaffolds for their potential as cytotoxic agents against a panel of human cancer cell lines. The compounds were subjected to the MTT cytotoxicity screening assay, which demonstrated that compounds 7d and 7e exhibited potent and selective cytotoxicity against HeLa and MCF-7 cell lines. These findings are promising as they offer a new avenue for the development of effective and targeted treatments for cancer. This study highlights the potential of thiophene-quinoline hybrids as a novel class of anticancer agents, worthy of further investigation59
 |
Figure 39 |
Vyas VK et al.(2019) generated and investigated a sequence of substituted quinoline derivatives with substitution at 2-, 4-, 6-, and/or 7th position as inhibitors of human dihydroorotate dehydrogenase (hDHODH) and potential anticancer agents in vitro. The substances were investigated for toxicity and docking score before being assessed for hDHODH inhibition and anticancer efficacy.
Compounds 7 and 14 demonstrated encouraging results against hDHODH, exhibiting IC50 values of 1.56 M and 1.22 M, respectively. In MTT assays, These compounds were also identified as having substantial anticancer action against with the types of cancer cells HT-29 and MDA-MB-231.The findings indicate that the quinoline derivatives produced in this study have the potential of being lead molecules in the generation of novel hDHODH inhibitors and anticancer agents. Overall, the findings provide a valuable contribution to the field of drug discovery and hold promise for the development of improved treatments for cancer60.
 |
Figure 40 |
Ruan et al. synthesized novel acridine and quinoline derivatives, which showed potent anti-proliferative activity against HepG-2 cells. Compound 3b displayed the highest activity with an IC50 of 261 nM, inhibited tubulin polymerization, disrupted microtubule dynamics, and induced cell cycle arrest in G2/M phase leading to cell apoptosis. Compound 3b also inhibited cancer cell migration in a dose-dependent manner. Docking studies revealed its fitting into the colchicine binding site of tubulin. These findings suggest that compound 3b is a promising tubulin inhibitor and warrants further investigation.61
 |
Figure 41 |
Conclusion
In conclusion, quinoline derivatives have shown remarkable potential as anticancer agents due to their diverse chemical structures and potent biological activity. The introduction of various functional groups and substitutions on the quinoline ring greatly influences the anticancer activity of the compound. Additionally, the ability of quinoline to combine with other heterocycles makes it a versatile scaffold for developing novel anticancer agents. The research on quinoline derivatives has demonstrated their effectiveness in inhibiting cancer cell growth, invasion, and migration, as well as inducing cell apoptosis. Hybrid derivatives of quinoline and other heterocycles have shown even greater activity against cancer cells, providing new avenues for developing improved treatments for cancer patients.
Moreover, the potential of quinoline as an extraordinary scaffold for anticancer agents must be further explored. Developing a perfect scaffold for anticancer agents remains a significant challenge, and optimization of various parameters such as selectivity, efficacy, pharmacokinetic properties, and toxicity is necessary to develop successful anticancer agents. By targeting specific signaling pathways and molecular targets, researchers can design quinoline derivatives that improve cancer treatment outcomes. Therefore, continued research on quinoline derivatives is essential to develop improved treatments for various malignancies, representing a significant step towards improving cancer treatments and drug discovery. Overall, the findings of this review highlight the potential of quinoline derivatives as versatile and effective anticancer agents and pave the way for further research in this area.
Acknowledgement
My heartfelt appreciation goes to Dr. A.K. Sheth, Head of the Pharmacy Department, and Dr. M.K. Mohan, Associate Professor in Pharmacognosy, for their invaluable guidance and support throughout this research.
Conflicts of Interest
We have no competing interests that could influence our research or findings; our work is based solely on scientific merit and integrity.
Funding Sources
There are no funding source.
References
- Afzal, O.; Kumar, S.; Haider, M. R.; Ali, M. R.; Kumar, R.; Jaggi, M.; Bawa, S. A Review on Anticancer Potential of Bioactive Heterocycle Quinoline. European Journal of Medicinal Chemistry. Elsevier Masson SAS 2015, pp 871–910. https://doi.org/10.1016/j.ejmech.2014.07.044.
- Tiwari A, Upmanyu N, C. A. A Text Book of Pharmaceutical Organic Chemistry-III,Nirali Prakashan, First.; Nirali prakashan, 2019.
- Tewari, N. Organic Chemistry: A Modern Approach, Volume III, First edit.; McGraw Hill Education(india) Private Limited, 2019.
- Quinoline _ C9H7N – PubChem.Pdf.
- Manske, R. H. F.; Kulka, M. The Skraup Synthesis of Quinolines. Org. React. 2011, 59–98. https://doi.org/10.1002/0471264180.OR007.02.
- Comprehensive Heterocyclic Chemistry III | ScienceDirect. https://www.sciencedirect.com/referencework/9780080449920/comprehensive-heterocyclic-chemistry-iii (accessed 2023-04-21).
- Kouznetsov, V.; Mendez, L.; Gomez, C. Recent Progress in the Synthesis of Quinolines. Curr. Org. Chem. 2005, 9 (2), 141–161. https://doi.org/10.2174/1385272053369196.
- Wang, Z. Comprehensive Organic Name Reactions and Reagents. Compr. Org. Name React. Reagents 2010. https://doi.org/10.1002/9780470638859.
- N, T. Organic Chemistry A Modern Approach Vol-III, First edit.; McGraw Hill Education(india) Private Limited, 2019.
- Tople, M. S.; Patel, N. B.; Patel, P. P. Microwave Irradiation for the Synthesis of Quinoline Scaffolds: A Review. J. Iran. Chem. Soc. 2023, 20 (1), 1–28. https://doi.org/10.1007/S13738-022-02648-Y/METRICS.
- Prajapati, S. M.; Patel, K. D.; Vekariya, R. H.; Panchal, S. N.; Patel, H. D. Recent Advances in the Synthesis of Quinolines: A Review. RSC Adv. 2014, 4 (47), 24463–24476. https://doi.org/10.1039/c4ra01814a.
- Beker, H. K.; Yıldırım, I. Anticancer Activity–Structure Relationship of Quinolinone-Core Compounds: An Overall Review. Pharm. Chem. J. 2023, 56 (10), 1333–1343. https://doi.org/10.1007/S11094-023-02794-4/METRICS.
- Ilakiyalakshmi, M.; Arumugam Napoleon, A. Review on Recent Development of Quinoline for Anticancer Activities. Arab. J. Chem. 2022, 15 (11), 104168. https://doi.org/10.1016/J.ARABJC.2022.104168.
- Jain, S.; Chandra, V.; Kumar Jain, P.; Pathak, K.; Pathak, D.; Vaidya, A. Comprehensive Review on Current Developments of Quinoline-Based Anticancer Agents. Arab. J. Chem. 2018, 12 (8), 4920–4946. https://doi.org/10.1016/j.arabjc.2016.10.009.
- Yadav, P.; Shah, K. Quinolines, a Perpetual, Multipurpose Scaffold in Medicinal Chemistry. Bioorg. Chem. 2021, 109, 104639. https://doi.org/10.1016/J.BIOORG.2021.104639.
- Moor, L. F. E.; Vasconcelos, T. R. A.; da R. Reis, R.; Pinto, L. S. S.; da Costa, T. M. Quinoline: An Attractive Scaffold in Drug Design. Mini Rev. Med. Chem. 2021, 21 (16), 2209–2226. https://doi.org/10.2174/1389557521666210210155908.
- Pimple, S.; Mishra, G. Cancer Cervix: Epidemiology and Disease Burden. Cytojournal 2022, 19. https://doi.org/10.25259/CMAS_03_02_2021.
- Formana, D.; de Martel, C.; Lacey, C. J.; Soerjomatarama, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; Franceschi, S. Global Burden of Human Papillomavirus and Related Diseases. Vaccine 2012, 30 (SUPPL.5). https://doi.org/10.1016/j.vaccine.2012.07.055.
- The Global Challenge of Cancer. Nat. Cancer 2020 11 2020, 1 (1), 1–2. https://doi.org/10.1038/s43018-019-0023-9.
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of Glucose Metabolization in Cancer Cells. J. Oncol. Sci. 2017, 3 (2), 45–51. https://doi.org/10.1016/J.JONS.2017.06.002.
- Bedard, P. L.; Hyman, D. M.; Davids, M. S.; Siu, L. L. Small Molecules, Big Impact: 20 Years of Targeted Therapy in Oncology. Lancet 2020, 395 (10229), 1078–1088. https://doi.org/10.1016/s0140-6736(20)30164-1.
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; Yang, S. Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives. Signal Transduct. Target. Ther. 2021 61 2021, 6 (1), 1–48. https://doi.org/10.1038/s41392-021-00572-w.
- Manfredi, S. The “Old” Cytotoxic Drugs, the Basis of Anti-Cancer Treatments. Therapies 2022, 77 (2), 251–255. https://doi.org/10.1016/J.THERAP.2021.11.006.
- Musiol, R. An Overview of Quinoline as a Privileged Scaffold in Cancer Drug Discovery. Expert Opin. Drug Discov. 2017, 12 (6), 583–597. https://doi.org/10.1080/17460441.2017.1319357.
- Mao, Y.; Soni, K.; Sangani, C.; Yao, Y. An Overview of Privileged Scaffold: Quinolines and Isoquinolines in Medicinal Chemistry as Anticancer Agents. Curr. Top. Med. Chem. 2020, 20 (28), 2599–2633. https://doi.org/10.2174/1568026620999200917154225.
- Matada, B. S.; Pattanashettar, R.; Yernale, N. G. A Comprehensive Review on the Biological Interest of Quinoline and Its Derivatives. Bioorg. Med. Chem. 2021, 32. https://doi.org/10.1016/J.BMC.2020.115973.
- Van de Walle, T.; Cools, L.; Mangelinckx, S.; D’hooghe, M. Recent Contributions of Quinolines to Antimalarial and Anticancer Drug Discovery Research. Eur. J. Med. Chem. 2021, 226. https://doi.org/10.1016/j.ejmech.2021.113865.
- R. Solomon, V.; Lee, H. Quinoline as a Privileged Scaffold in Cancer Drug Discovery. Curr. Med. Chem. 2011, 18 (10), 1488–1508. https://doi.org/10.2174/092986711795328382.
- Kaschula, C. H.; Egan, T. J.; Hunter, R.; Basilico, N.; Parapini, S.; Taramelli, D.; Pasini, E.; Monti, D. Structure-Activity Relationships in 4-Aminoquinoline Antiplasmodials. The Role of the Group at the 7-Position. J. Med. Chem. 2002, 45 (16), 3531–3539. https://doi.org/10.1021/JM020858U.
- Srivastava, S. K.; Jha, A.; Agarwal, S. K.; Mukherjee, R.; Burman, A. C. Synthesis and Structure-Activity Relationships of Potent Antitumor Active Quinoline and Naphthyridine Derivatives. Anticancer. Agents Med. Chem. 2007, 7 (6), 685–709. https://doi.org/10.2174/187152007784111313.
- Mohamed, M. F. A.; Abuo-Rahma, G. E. D. A. Molecular Targets and Anticancer Activity of Quinoline–Chalcone Hybrids: Literature Review. RSC Adv. 2020, 10 (52), 31139–31155. https://doi.org/10.1039/D0RA05594H.
- Marella, A.; Tanwar, O. P.; Saha, R.; Ali, M. R.; Srivastava, S.; Akhter, M.; Shaquiquzzaman, M.; Alam, M. M. Quinoline: A Versatile Heterocyclic. Saudi Pharm. J. 2013, 21 (1), 1–12. https://doi.org/10.1016/J.JSPS.2012.03.002.
- Narwal, S.; Kumar, S.; Verma, P. K. Synthesis and Therapeutic Potential of Quinoline Derivatives. Res. Chem. Intermed. 2017, 43 (5), 2765–2798. https://doi.org/10.1007/S11164-016-2794-2/METRICS.
- Zhou, P.; Aschauer, U.; Decurtins, S.; Feurer, T.; Häner, R.; Liu, S. X. Effect of Tert -Butyl Groups on Electronic Communication between Redox Units in Tetrathiafulvalene-Tetraazapyrene Triads. Chem. Commun. 2021, 57 (96), 12972–12975. https://doi.org/10.1039/D1CC05671A.
- Assefa, H.; Kamath, S.; Buolamwini, J. K. 3D-QSAR and Docking Studies on 4-Anilinoquinazoline and 4-Anilinoquinoline Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors. J. Comput. Aided. Mol. Des. 2003, 17 (8), 475–493. https://doi.org/10.1023/B:JCAM.0000004622.13865.4f.
- Chen, Y. L.; Hung, H. M.; Lu, C. M.; Li, K. C.; Tzeng, C. C. Synthesis and Anticancer Evaluation of Certain Indolo[2,3-b]Quinoline Derivatives. Bioorganic Med. Chem. 2004, 12 (24), 6539–6546. https://doi.org/10.1016/j.bmc.2004.09.025.
- Tseng, C. H.; Chen, Y. L.; Yang, C. L.; Cheng, C. M.; Han, C. H.; Tzeng, C. C. Synthesis of 6-Substituted 9-Methoxy-11H-Indeno[1,2-c]Quinoline-11-One Derivatives as Potential Anticancer Agents. Bioorganic Med. Chem. 2012, 20 (14), 4397–4404. https://doi.org/10.1016/j.bmc.2012.05.035.
- Ghorab, M. M.; Ragab, F. A.; Noaman, E.; Heiba, H. I.; El-Hossary, E. M. Synthesis of Some Novel Quinolines and Pyrimido[4,5-b]Quinolines Bearing A Sulfonamide Moiety as Potential Anticancer and Radioprotective Agents; 2007; Vol. 57.
- Scott, D. A.; Balliet, C. L.; Cook, D. J.; Davies, A. M.; Gero, T. W.; Omer, C. A.; Poondru, S.; Theoclitou, M. E.; Tyurin, B.; Zinda, M. J. Identification of 3-Amido-4-Anilinoquinolines as Potent and Selective Inhibitors of CSF-1R Kinase. Bioorganic Med. Chem. Lett. 2009, 19 (3), 697–700. https://doi.org/10.1016/j.bmcl.2008.12.046.
- Mai, A.; Rotili, D.; Tarantino, D.; Nebbioso, A.; Castellano, S.; Sbardella, G.; Tini, M.; Altucci, L. Identification of 4-Hydroxyquinolines Inhibitors of P300/CBP Histone Acetyltransferases. Bioorganic Med. Chem. Lett. 2009, 19 (4), 1132–1135. https://doi.org/10.1016/j.bmcl.2008.12.097.
- Miller, L. M.; Mayer, S. C.; Berger, D. M.; Boschelli, D. H.; Boschelli, F.; Di, L.; Du, X.; Dutia, M.; Floyd, M. B.; Johnson, M.; Kenny, C. H.; Krishnamurthy, G.; Moy, F.; Petusky, S.; Tkach, D.; Torres, N.; Wu, B.; Xu, W. Lead Identification to Generate 3-Cyanoquinoline Inhibitors of Insulin-like Growth Factor Receptor (IGF-1R) for Potential Use in Cancer Treatment. Bioorganic Med. Chem. Lett. 2009, 19 (1), 62–66. https://doi.org/10.1016/j.bmcl.2008.11.037.
- Chen, S.; Chen, R.; He, M.; Pang, R.; Tan, Z.; Yang, M. Design, Synthesis, and Biological Evaluation of Novel Quinoline Derivatives as HIV-1 Tat-TAR Interaction Inhibitors. Bioorganic Med. Chem. 2009, 17 (5), 1948–1956. https://doi.org/10.1016/j.bmc.2009.01.038.
- Ghorab, M. M.; Ragab, F. A.; Heiba, H. I.; Arafa, R. K.; El-Hossary, E. M. In Vitro Anticancer Screening and Radiosensitizing Evaluation of Some New Quinolines and Pyrimido[4,5-b]Quinolines Bearing a Sulfonamide Moiety. Eur. J. Med. Chem. 2010, 45 (9), 3677–3684. https://doi.org/10.1016/j.ejmech.2010.05.014.
- Wang, Y.; Ai, J.; Wang, Y.; Chen, Y.; Wang, L.; Liu, G.; Geng, M.; Zhang, A. Synthesis and C-Met Kinase Inhibition of 3,5-Disubstituted and 3,5,7-Trisubstituted Quinolines: Identification of 3-(4-Acetylpiperazin-1-Yl)-5- (3-Nitrobenzylamino)-7- (Trifluoromethyl)Quinoline as a Novel Anticancer Agent. J. Med. Chem. 2011, 54 (7), 2127–2142. https://doi.org/10.1021/jm101340q.
- Patel, R. V.; Kumari, P.; Rajani, D. P.; Chikhalia, K. H. Synthesis and Studies of Novel 2-(4-Cyano-3-Trifluoromethylphenyl Amino)-4-(Quinoline-4-Yloxy)-6-(Piperazinyl/Piperidinyl)-s-Triazines as Potential Antimicrobial, Antimycobacterial and Anticancer Agents. Eur. J. Med. Chem. 2011, 46 (9), 4354–4365. https://doi.org/10.1016/j.ejmech.2011.07.006.
- Tseng, C. H.; Chen, Y. L.; Yang, C. L.; Cheng, C. M.; Han, C. H.; Tzeng, C. C. Synthesis of 6-Substituted 9-Methoxy-11H-Indeno[1,2-c]Quinoline-11-One Derivatives as Potential Anticancer Agents. Bioorganic Med. Chem. 2012, 20 (14), 4397–4404. https://doi.org/10.1016/j.bmc.2012.05.035.
- Ai, Y.; Liang, Y. J.; Liu, J. C.; He, H. W.; Chen, Y.; Tang, C.; Yang, G. Z.; Fu, L. W. Synthesis and in Vitro Antiproliferative Evaluation of Pyrimido[5,4-c] Quinoline-4-(3H)-One Derivatives. Eur. J. Med. Chem. 2012, 47 (1), 206–213. https://doi.org/10.1016/j.ejmech.2011.10.044.
- Marganakop, S. B.; Kamble, R. R.; Taj, T.; Kariduraganvar, M. Y. An Efficient One-Pot Cyclization of Quinoline Thiosemicarbazones to Quinolines Derivatized with 1,3,4-Thiadiazole as Anticancer and Anti-Tubercular Agents. Med. Chem. Res. 2012, 21 (2), 185–191. https://doi.org/10.1007/s00044-010-9522-z.
- Kouznetsov, V. V; Rojas Ruíz, F. A.; Vargas Méndez, L. Y.; Gupta, M. P. Simple C-2-Substituted Quinolines and Their Anticancer Activity; 2012; Vol. 9.
- Ökten, S.; Çakmak, O.; Erenler, R.; Yüce, Ö.; Tekin, Ş. Simple and Convenient Preparation of Novel 6,8-Disubstituted Quinoline Derivatives and Their Promising Anticancer Activities. Turkish J. Chem. 2013, 37 (6), 896–908. https://doi.org/10.3906/kim-1301-30.
- Al-Dosari, M. S.; Ghorab, M. M.; Alsaid, M. S.; Nissan, Y. M.; Ahmed, A. B. Synthesis and Anticancer Activity of Some Novel Trifluoromethylquinolines Carrying a Biologically Active Benzenesulfonamide Moiety. Eur. J. Med. Chem. 2013, 69, 373–383. https://doi.org/10.1016/j.ejmech.2013.08.048.
- Sun, J.; Zhu, H.; Yang, Z. M.; Zhu, H. L. Synthesis, Molecular Modeling and Biological Evaluation of 2-Aminomethyl-5-(Quinolin-2-Yl)-1,3,4-Oxadiazole-2(3H)-Thione Quinolone Derivatives as Novel Anticancer Agent. Eur. J. Med. Chem. 2013, 60, 23–28. https://doi.org/10.1016/j.ejmech.2012.11.039.
- Badowska-Roslonek, K.; Godlewska, J.; Switalska, M.; Piskozub, M.; Peczynska-Czoch, W.; Wietrzyk, J.; Kaczmarek, L. NEW 6H-INDOLO[2,3-B]QUINOLINE O-AMINOGLYCOSIDES OVERCOMING ANTICANCER MULTIDRUG RESISTANCE; Vol. 7. www.arpapress.com/Volumes/JPCS/Vol7/JPCS_7_02.pdf.
- Makawana, J. A.; Sangani, C. B.; Lin, L.; Zhu, H. L. Schiff’s Base Derivatives Bearing Nitroimidazole and Quinoline Nuclei: New Class of Anticancer Agents and Potential EGFR Tyrosine Kinase Inhibitors. Bioorganic Med. Chem. Lett. 2014, 24 (7), 1734–1736. https://doi.org/10.1016/j.bmcl.2014.02.041.
- Vyas, V. K.; Variya, B.; Ghate, M. D. Design, Synthesis and Pharmacological Evaluation of Novel Substituted Quinoline-2-Carboxamide Derivatives as Human Dihydroorotate Dehydrogenase (HDHODH) Inhibitors and Anticancer Agents. Eur. J. Med. Chem. 2014, 82, 385–393. https://doi.org/10.1016/j.ejmech.2014.05.064.
- Ahsan, M. J.; Shastri, S.; Yadav, R.; Hassan, M. Z.; Bakht, M. A.; Jadav, S. S.; Yasmin, S. Synthesis and Antiproliferative Activity of Some Quinoline and Oxadiazole Derivatives. Org. Chem. Int. 2016, 2016, 1–10. https://doi.org/10.1155/2016/9589517.
- Shaikh, S. K. J.; Kamble, R. R.; Somagond, S. M.; Devarajegowda, H. C.; Dixit, S. R.; Joshi, S. D. Tetrazolylmethyl Quinolines: Design, Docking Studies, Synthesis, Anticancer and Antifungal Analyses. Eur. J. Med. Chem. 2017, 128, 258–273. https://doi.org/10.1016/j.ejmech.2017.01.043.
- George, R. F.; Samir, E. M.; Abdelhamed, M. N.; Abdel-Aziz, H. A.; Abbas, S. E. S. Synthesis and Anti-Proliferative Activity of Some New Quinoline Based 4,5-Dihydropyrazoles and Their Thiazole Hybrids as EGFR Inhibitors. Bioorg. Chem. 2019, 83, 186–197. https://doi.org/10.1016/j.bioorg.2018.10.038.
- Othman, D. I. A.; Selim, K. B.; El-Sayed, M. A. A.; Tantawy, A. S.; Amen, Y.; Shimizu, K.; Okauchi, T.; Kitamura, M. Design, Synthesis and Anticancer Evaluation of New Substituted Thiophene-Quinoline Derivatives. Bioorganic Med. Chem. 2019, 27 (19). https://doi.org/10.1016/j.bmc.2019.07.042.
- Vyas, V. K.; Qureshi, G.; Oza, D.; Patel, H.; Parmar, K.; Patel, P.; Ghate, M. D. Synthesis of 2-,4,-6-, and/or 7-Substituted Quinoline Derivatives as Human Dihydroorotate Dehydrogenase (HDHODH) Inhibitors and Anticancer Agents: 3D QSAR-Assisted Design. Bioorganic Med. Chem. Lett. 2019, 29 (7), 917–922. https://doi.org/10.1016/j.bmcl.2019.01.038.
- Ren, Y.; Ruan, Y.; Cheng, B.; Li, L.; Liu, J.; Fang, Y.; Chen, J. Design, Synthesis and Biological Evaluation of Novel Acridine and Quinoline Derivatives as Tubulin Polymerization Inhibitors with Anticancer Activities. Bioorg. Med. Chem. 2021, 46, 116376. https://doi.org/10.1016/J.BMC.2021.116376.

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


