Synthesis of Salicylate and Salicylamide Alcohols for the Preparation of Phosphorodiamidates and Ifosfamide Prodrugs.
Ashutosh Pal
Department of Chemistry, Raja Peary Mohan College, West Bengal, India.
Corresponding Author E-mail: ashupal33@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/370205
Article Received on : 12-Jan-2021
Article Accepted on :
Article Published : 30 Mar 2021
Pro drugs are derivatives of drug substance which gives parent drug or release drug when it breaks inside the body by the presence of suitable enzyme,and then exert desired pharmacological effect. For many years, prodrug strategy has been developed enormously to solve many unwanted drug properties. In drug discovery and development, prodrugsare well-known pharmacokinetic effects of pharmacologically nimbleproducts. Almost10% of drugs permitted whole worldare classified as prodrugs, where the application of a prodrug method duringinitial stages of drug development is an emergent fashion. Phosphorodiamidates prodrugs are well known anticancer agents particularly against leucomia. To improve the selectivity of the chemotherapeutic agents and reduce systemic toxicity, I herein report different types of salicylate and salicylamide alcohols for the preparation of phosphorodiamidates and ifosfamide prodrugs.
KEYWORDS:Drugs; Ifosfamide Prodrugs; Prodrugs; Phosphorodiamidates; Suitable Enzyme; Salicylate and Salicylamide Alcohols
Download this article as:| Copy the following to cite this article: Pal A. Synthesis of Salicylate and Salicylamide Alcohols for the Preparation of Phosphorodiamidates and Ifosfamide Prodrugs. Orient J Chem 2021;37(2). |
| Copy the following to cite this URL: Pal A. Synthesis of Salicylate and Salicylamide Alcohols for the Preparation of Phosphorodiamidates and Ifosfamide Prodrugs. Orient J Chem 2021;37(2). Available from: https://bit.ly/3m0nEMw |
Introduction
Prodrugs are precursors of drug substanceswhich are pharmacologically inactive it necessary either enzymatic alteration or chemically transformation to the main drug in vivo so order as to achieve a pharmacological consequence. Prodrug is more potent than parent drug just like nascent hydrogen. Sometimes parent drug molecule becomes less delivery properties than its prodrugmolecule1. The concept of prodrug is justified due to it allow the proper functioningdrug to surpass betterof barrier that would obstruct it from reaching the site of action to exercise the necessary pharmacological activity.
At present due to unwanted side effects of maximum drugs like poor bioavailability, incomplete absorption, short period of action, non-specificity, organoleptic properties, less solubility in water, high first-pass metabolism or other adversarial effects (propranolol); short half-life arise due to metabolic instability, bad permeability or absorption (ampicillin); (dopamine); site specificity is not properly (anticancer agents); unfriendly organoleptic properties (chloramphenicol); incomplete absorption (epinephrine); cause difficult during formulation and disadvantageous effects and toxicity2-4that hamper their therapeutic effectiveness.
In the maneuver of drug delivery processthe prodrug approach is quicklytaking a crucial part during treatment of patient. The implementation of prodrug strategy in the past30 years has accelerated a firm progress in the biopharmaceutical, physicochemical and/or pharmacokinetic attributes of the pharmacologically active substances. It is measured the success of the prodrug approach from the survey that thenumber of prodrugs are presently on the market. About 10 to 15 % of promoted drugs can be categorized as prodrugs now, and up to year 2008, 30 % of approved minor molecular weight drugs were prodrugs5-7. During the period of 2008–2017, 12% of drugs molecules approved by the FDA were prodrugs8-10.
Though enormous advances have occurred in the field of cancer, it still remains a major health problem and it has been reported that cancer is the cause of death up to 25 % in USA. At the present time, cyclophosphamide (CPA) is the most frequently used agent of the alkylating agent class in medical oncology11-17. Two congeners, ifosfamide (IPM) and trotosfamide are also in clinical cancertreatment. CPA has better therapeutics index (particularly in the treatment of ovarian and breast cancer) than other mustard drugs like nitrogen mustard and exhibit broad area of clinical efficacythough the main biotransformation pathway of phosphamide drug is well designed, its appliance of tumoridical selectivity remains controversial18-22.
This article presents two types alcohols on which first type approach is ester form so that the prodrug can cleave by the presence of esterase and release ifosphoramide mustard with physically healthy compound salicylic acid and the second type that is amide form should fail to cleave by the esterase enzyme because these are amide derivatives which should not cleaved and release drug by the presence of esterase.
Chemistry
The synthesis of the target alcohols was carried out as shown in Scheme 2 to 4 and phosphorodiamidates can be easily synthesized by the reaction of alcohol and phosphorous oxychloride followed by the treatment of chloroethylamine hydrochloride as in Scheme 115.
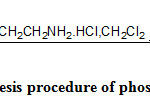 |
Scheme 1: Synthesis procedure of phosphorodiamidates. |
Compounds 4a-b, 10 and 14a-b those have not been reported previously, were prepared by stirring with the mixture of phenyl salicylate and the corresponding aldehyde23using DABCO as base in absence of solvent24 as shown in Scheme 2.
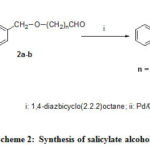 |
Scheme 2: Synthesis of salicylate alcohols. |
The synthetic approach to 10 is based on 6, and 7. But in that case aldehyde was nitro substituted and salicylate was 4-benzyloxy substituted (Scheme 3). Molecules14a-b were generated from N-methyl salicylamide and the corresponding alcohol in presence of ortho phosphoric acid in tetrahydofuran at refluxing temperature 25 shown in Scheme 4. Hydrogenelysis of 3a-b and 13a-bwere carried out over 10% Pd/C under pressure using ethyl acetate as solvent and the trace of 70% perchloric acid as catalyst yielded corresponding alcohols. Alcohol 10 was synthesized by photolysis26 from compound 9.
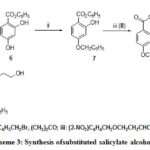 |
Scheme 3: Synthesis ofsubstituted salicylate alcohols. |
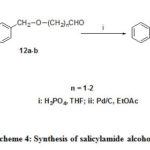 |
Scheme 4: Synthesis of salicylamide alcohols. |
Cyclic acetal phosphorodiamidatesderivatives were synthesized as the test reaction to confirm the formation of mustard using those alcohols. Alcohols 4a-b were converted into phosphorodichloridates by POCl3and Et3N in dichloromethane at – 20oC which in situ, were converted to phosphorodiamidates by the treatment with 2-fold molar equivalent of 2-chloroethylamine hydrochloride Scheme-1.
Experimental Section
All NMR spectra were drawn from IBM-Brucker Model NR/200 AF spectrometer in the FT model, in deutero chloro form where tetramethyl sillaneused as an internal standard. The chemical shift represents δppm and coupling constant represents J in hertz and using by Hoover capillary apparatus melting points of the compounds were checked. Reactions in this article were carried out in dry glass apparatus and inert atmosphere using nitrogen and helium as inert gas. Dry and analytical solvents were used for all reactions. The reaction progress and homogeneity of the reaction mixture were checked by TLC coated with silica gel that was run in glass plates using the following solvent mixtures (a), CHCl3/MeOH (19:1 to 9.5:0.5 v/v); (b), hexane/EtOAc (9:1 to 1:1); and (c), 100% diethyl ether. TLC plates were visualized under a UV lamp (254nm) and spraying agent used as anisaldehyde solution in 95% ethanol and heated to 100 oC for 5 min appeared as different coloured spots. Pure products were obtained by chromatography with neutral alumina or on silica gel with the solvent mixture of hexane/EtOAc or CH2Cl2/MeOH. The reactions mixture were extracted with diethyl ether, ethyl acetate, dichloromethane or trichlorome than and ranched with aqua, brine solution, saturated sodium bicarbonate whenever necessary and then dried with anhydrous MgSO4. Solutions were concentrated using rotary evaporator under reduced pressure and dried.
3-Benzyloxypropionaldehyde (2a).
In amixture of phenyl methanol (61.5 mL, 64.25 g, 0.595 mol), 2-chloroethanoic acid (3.36 g),and NaOH (1.425 g) in 7.50 mL water added slowly with gentle shaking for 15 min to acroline (50 mL, 42.95 g, 0.75 mol). At 40 oC 15 mL aceteic acid drop wise added to the reaction system and maintained it at 40 oC during 80 hours. The crude reaction mixture was worked up with EtOAc and acid was removed by wash with aqua (75 mL x 3) and passed throughanhydrous MgSO4. After work up the organic solvent was dried and crude product made pure by vacuum distillation at 100 oC (0.3 mmHg) pressure followed by the removal of volatile starting materials and side products. The residue remained as viscous oil was aldehyde 2a confirmed from NMR studies. The raw material was taken for subsequent stepsand there is no necessary for purication.Crude yield was 33 g ( 32%). Proton NMR values were determined by deuterium chloroform;Proton-NMR δ: 9.78 (-CHO), 7.29 (Ar-H), 4.52 (C6H5CH2), 3.80 (-O-CH2), 2.68 (-CH2); C-13NMR (CDCl3) δ : 200.87, 137.70, 127.50 and 127.34 (aromatic), 72.92 (benzylic), 66.57, 43.58; MS (C9H10O2) calcd. 150.068 found MH+151.1.
2-(2´-Benzyloxyethyl)-4H-1,3-benzdioxin-4-one (3a).
This reaction was performed with a mixture DABCO (1.1g, 10 mmol), (2.15 g) phenyl salicylateand (1.518 g) of 3-benzyloxypropionaldehyde without solvent and was warmed to 40 oC during 12 minutes, then it continued during overnight at normal warmth condition where suspension formed which was extracted by EtOAc and was washed thoroughly with 30 mL 2% NaOH solution two times and aqua (30 mL x2) and passed through drying agent. The crude compound was achieved after removal of organic solvent which made pure by chromatography (ca. 200 g) taking hexane/EtOAc (98:2, v/v) as solvent mixture to yield 3a clear liquid form (880mg, 68%): Rf 0.55 (95:5, hexane/EtOAc). Proton NMR δ : 7.98 (Ar-H), 7.55 (Ar-H), 7.30 (Ar-H)), 7.16 (Ar-H), 7.00 (Ar-H)), 5.81 (-O-CH-O-), 4.52 (C6H5CH2), 3.76 (-O-CH2-CH2-), 2.35 (-O-CH2-CH2-).Mass spectroscopy (C16H14O4) calcd. 270.089 found MH+271.2.
2-(2´-Hydroxyethyl)-4H-1,3-benzdioxin-4-one (4a).
(1.0 g, 3.52 mmol) 2-(2´-benzyloxyethyl)-4H-1,3-benzdioxin-4-one 3a in 10 mL ethyl acetate containing 10 μL of percholoric acid (70%) was reduced with gaseous hydrogen with150 mg 10% palladium on carbon under pressure during 25 min. Percholoric acid was then neutralized with finely powdered 800 mg of CaCO3. After the reaction it was filtered through glass wool and dried, theresidue was got pureafter chromatography using hexane-ethyl acetate (80:20) mixture to yield 4a as oil (628 mg, 92%).Proton-NMR (CDCl3) δ : 7.94 (Ar-H), 7.56 (Ar-H), 7.16 (Ar-H), 7.04 (Ar-H), 5.83 (-O-CH-O-), 3.95 (-O-CH2-CH2-), 2.89 (bs, 1H, OH), 2.30 ((-O-CH2-CH2-); C-13 NMR δ : 162.24, 158.16, 136.22, 130.01, 123.33, 116.66, 114.22, 99.73, 56.71, 36.21; MS (C9H8O4) calcd. 180.042 found MH+181.1.
4-Benzyloxy-1-butanal (2b).
Butane-1,4-diol (5.91 g, 66mmol) and powdered potassium hydroxide (1.78 g, 32 mmol) was warmedat 120 oC and after that the diol made free from water by vacuum distillation. Benzyl bromide (4.44 g, 26 mmol) was then added slowly at 100 oC from the dropping funnel. The reaction was continued to stirr for another 2 h at 100 oC.The product was stirred with 20 mL water after cooling it at room temperature.The crude product was extracted and dried with anhydrous MgSO4, concentrated and distilled to give 9.57 g (81%) of the mono protected alcohol (4-benzyloxy-1-butanol).
4-Benzyloxy-1-butanol (3.89 g) was taken in a dropping funnel and added slowly to a mixtute of PCC (7.28 g) and dryCH3COONa (30 mg) dissolved in 50 mL of dichloromethane and stirred at room temperature for 2 h and thesolution was rapidly passed through a small silica gel column. The filtrate part was concentrated and distilled quickly to give 4-benzyloxy-1-butanal as oil, 2.62 g (68%). Proton-NMR (CDCl3) δ : 9.78 (-CHO), 7.30 (Ar-H), 4.49 (s, 1H, benzylic), 3.35 (-O-CH2), 2.55 (-O-CH2-CH2), 1.95 (C-3H). C-13 NMR (CDCl3)δ : 202.0, 128.35, 128.30. 127.80 and 127.50 (aromatic), 72.90 (benzylic) , 69.10 , 41.00, 22.60. MS (C10H12O2) calcd. 164.084 found MH+165.1.
2-(3´-Benzyloxypropyl)-4H-benz-1,3-dioxin-4-one (3b).
Compound 2b wasconverted into3bas described for the synthesis of 3a (5.76 mmol scale except benzyloxypropanaldehyde was used as aldehyde). After complete the reaction and purification the product benzodioxinone 3b was obtained, as liquid (1.13 g, 67%). 1H NMR δ : 7.97 (Ar-H), 7.54 (Ar-H), 7.32 (Ar-H), 7.15(Ar-H), 7.01(Ar-H), 5.64 (-O-CH-O-), 4.52 (s, 2H, benzylic), 3.57 (-O-CH2), 2.15 (O-CH2-CH2),), 1.92 (O-CH2-CH2),); MS (C17H16O4) calcd. 284.105 found MH+285.2.
2-(3´-Hydroxypropyl)-4H-benz-1,3-dioxin-4-one (4b).
Compound 2-(3´-benzyloxypropyl)-4H-benz-1,3-dioxin-4-one (2.0 g) was taken in20mL ethyl acetate containing 10 μL of percholoric acid (70%) was reduced with gaseous hydrogen with150 mg 10% palladium on carbon under pressure during 25 min same as the transformation from 3a to 4a. Solvent was dried and concentratedusing pump (0.03 mm Hg) to yield 4b (1.37 g, 94%) as a clear oil. The colourless oily substance was crystallized on storage at –13 oC. Mp: 44 oC. Proton- NMR δ : 7.99 (Ar-H), 7.57 (Ar-H), 7.18 (Ar-H), 7.04 (Ar-H), 5.69 (-O-CH-O-), 3.65 (-O-CH2), 2.21(O-CH-O-), 2.11 (-O-CH-O-). C-13 NMR (CDCl3)δ: 162.76, 158.76, 136.66, 130.59, 123.77, 117.04 and 114.79 (aromatic), 101.82, 62.33, 30.53, 26.33; MS (C10H10O4) calcd. 194.058 found MH+195.2.
2,4-Dihydroxyphenylbenzoate (6).
Phenol (6.1 g, .065 mol), 2,4-dihydroxybenzoic acid (11 g) andtrifluoroacetic anhydride (20 g) were mixed in ether (50 mL) and it wash eated to refluxed at 65 oC for 2 h. 200 mL ether was added to the solution and ether layer was shaken well with saturated bicarbonate, brine solutions and concentrated. The crude product was treated with Norit in toluene and filtered. Hexane was added to it to get a white powder that was free of phenol. The compound was purified by crystalization from toluene-hexane mixture (6:4, v/v) provided white prisms (10.2 g, 67%). Mp: 147-149 oC. Proton-NMR (CDCl3) δ : 10.73 (OH at C-2), 7.95 (Ar-H), 7.43 (Ar-H), 7.28 (Ar-H), 7.18 (Ar-H), 6.43 (m, 2H), 6.09 (bs, 1H, OH); MS (C13H10O4) calcd. 230.058 found MH+231.2.
Phenyl-2-hydroxy-4-benyloxybenzoate (7).
A solution of phenyl-2,4-dihydroxybenzoate (1 g), benzyl bromide (744 mg), K2CO3 (684 mg) in 30 mL acetone was heated to refluxed for a day and night. Acetone was removed from the reaction mixture andthe crude product was work-up with CHCl3, washed with 10% (v/v) HCl/H2O and then brine, passed through anhydrous MgSO4 and evaporated. The pure white solid was obtained by flash chromatography yielded phenyl-2-hydroxy-4-benzyloxybenzoate (0.64 g, 46%). Mp: 108-109 oC. Proton- NMR (CDCl3) δ : 10.69 (OH at C-2), 7.97 (Ar-H), 7.29 (Ar-H), 6.59 (Ar-H), 5.10 (s, benzylic); MS (C20H16O4) calcd. 320.105 found MH+321.3.
2-nitrobenzyl alcohol (11.0 g, 0.072 mol),chloroacetic acid (1.68 g) and NaOH (0.713 g) in water (4 mL)was added slowlyfrom dropping funnel with stirring over 20 min to acrolein (25 mL, 20.98 g).The solution was acidified with CH3COOH (7.5 mL, 0.13 mol) and warmed at 40 oC for 80 h. The solution was chilled, shaken with water (40×3 mL), and dried. The crude residue was then purified by chromatography furnished yellow oily product (7 g, 46.6 %). Proton NMR (CDCl3) δ : 9.83 (-CHO), 8.04 (Ar-H), 7.74 (Ar-H), 7.64 (Ar-H), 7.44 (Ar-H), 4.90 (s, 2H, benzylic), 3.93 (-O-CH-O-), 2.76 (m, -O-CH2-CH2). C-13 NMR (CDCl3) δ : 201.1, 147.6, 134.9, 134.0, 129.0, 128.5, 125.0, 70.0, 65.1, 44.1; MS (C10H11NO4) calcd. 209.069 found MH+210.2.
2-[2´-(o-Nitrobenzyloxyethyl)-4H-1,3-benzdioxin-4-one (9).
A mixture of 3-(2´-nitrobenzyloxy)propionaldehyde (670mg, 3.2 mmol), 4-benzyloxy-2-hydroxy phenyl benzoate (1.1 g)and DABCO (380mg) was warmed 40 oC for 20 min and stirred itduring 72 h at rt without solvent. Reaction mixture was then extracted with ethyl acetate. The organic layer was washed with 2% NaOH solution (30x3mL), then with water and concentrated. Pure product thenobtained by flash chromatography to yield9(610 mg, 41%) as a viscuss oily substance.Proton NMR (CDCl3) δ : 8.03 (Ar-H), 7.89 (Ar-H), 7.73 (Ar-H), 7.61 (Ar-H), 7.39 (Ar-H), 6.77 (Ar-H), 6.57 (Ar-H), 5.80 (Ar-H), 5.11 (s, CH2C6H5), 4.90 (s, 2H, NO2C6H4CH2), 3.85 (-O-CH-O-), 2.37 (q, 2H, -O-CH2-CH2).C-13 NMR (75 MHz, CDCl3): δ 165.2 (C-4´), 161.9, 160.2, 147.3, 135.6, 134.6, 133.6, 131.8, 128.7, 128.7, 128.6, 128.4, 128.1, 127.4, 127.4, 124.6, 111.9, 107.3 and 101.3 (aromatic) 99.3 , 70.5 and 69.7 (benzylic), 65.2 and 34.1 (methylene); MS (C24H21NO7) calcd. 435.132 found MH+436.3.
2-(2´-Hydroxyethyl)-4H-7-benzyloxy-1,3-benzdioxin-4-one (10).
In a 200 mL rb was charged with 9 (300 mg, mmol), in 35mL of dry THF. Reaction vessel was then purged with argon duringhalf an hour and was stirred by keeping outside in ACE photochemical UV power supplies and murcurry vapour lamp for 7 h. The solution was concentrated after the reaction yielded dark orange-red oil. The crude alcohol was purified by chromatograpy to yieldwhite crystalline solid 160 mg (77%), Mp: 110 –111 oC. Proton- NMR (CDCl3) δ : 7.85 (Ar-H), 7.36 (Ar-H), 6.75 (Ar- H), 6.55 (Ar-H), 5.78 (Ar-H), 5.07 (Ar-H), 3.92 (-O-CH-O-), 2.56 (bs, 1H, OH ), 2.26 (q, 2H,-O-CH2-CH2). C-13 NMR (CDCl3) δ : 165.6 (C-4), 162.6, 160.6, 135.9, 132.2, 129.2, 129.2, 128.8, 127.9, 127.9, 112.4, 107.5 and 101.8 (aromatic), 100.3, 70.9 (benzylic), 57.4 , 36.8; MS (C17H16O5) calcd. 300.100 found MH+301.1.
2-(2´-Benzyloxyethyl)-3-methyl-4H-1,3-benzoxazin-4-on (13a).
Benzyloxy- propanaldehyde (4.5 g, 0.027 mol) and5 mL ortho-phosphoric acid was mixed N-methyl salicylamide (4.14 g, 0.027 mol) in THF. Thereaction vessel was refluxed for 14 h (TLC control). Then THF was removed from the system andwork up with ethyl acetate and concentrated. The pure product was obtained by flash chromatographed to give 13a as anoily substance (2.4 g, 31.6%). Proton NMR (CDCl3) δ : 7.93 (Ar-H), 7.33 (Ar-H), 7.02 (Ar-H), 6.86 (Ar-H), 5.49 (Ar-H), 4.49 (s, 2H, benzylic), 3.54 (-O-CH-O-), 3.03 (s, N-CH3), 2.08 (m, O-CH2-CH2); MS (C17H17NO3) calcd. 283.121 found MH+284.3.
2-(2´-Hydroxyethyl)-3-methyl-4H-1,3-benzoxazin-4-one (14a).
Substance 2-(2´-benzyloxyethyl)-4H-1,3-benzoxazin-4-one 13a, was converted to 14a as reported for the synthesis of 13a (1.5 mmol scale). The pure compound was obtained by flash chromatography to yield 14a as an oily substance (300 mg, 96%), Proton NMR (CDCl3) δ : 7.93 (Ar-H), 7.42 (Ar- H), 7.08 (Ar-H), 6.9 (Ar-H), 5.53 (Ar-H), 3.8 (m, -O-CH-O-), 3.10 (s, 3H, NCH3 ), 2.1 (m, -O-CH2-CH2); C-NMR NMR (CDCl3): δ 162.2, 155.7, 134.72, 127.5, 122.5, 118.2, 117.2, 87.0, 57.1, 34.5, 30.5 (NCH3); MS (C10H11NO3) calcd. 193.074 found MH+194.2.
2-(3´-Benzyloxypropyl)-3-methyl-4H-benz-1,3-dioxin-4-one (13b).
Product 13b was prepared from 12b as described for the synthesis of 13a (13.2 mmol scale except benzyloxy- propanaldehyde was used as aldehyde). The pure compound was obtained by flash chromatography to efford the benzodioxinone 13b, as an oily substance (1.4 g, 37%). Rf = 0.45 (hexane/EtOAc, 6.5 : 3.5). Proton NMR (CDCl3) δ : 7.90 (Ar-H), 7.31 (Ar-H), 6.97 (Ar-H), 7.15 (Ar-H), 6.85 (Ar-H), 5.26 (Ar-H), 4.47 (s, 2H, benzylic), 3.50 (m, -O-CH-O-), 3.05 (s, 3H, NCH3), 1.85 (m, -O-CH2-CH2-), MS (C18H19NO3) calcd. 297.137 found MH+298.3.
2-(2´-Hydroxypropyl)-3-methyl-4H-1,3-benzoxazin-4-one (14b).
2-(2´-benzyloxypropyl)-4H-1,3-benzoxazin-4-one 13b, was converted to 14b as mentioned for the synthesis of 13a (1.5 mmol scale). The pure compound was obtained by flash column chromatography on silica using CHCl3/MeOH (97:3 to 95:5, v/v) as eluent to afford 14b as a colorless oil (310 mg, 97% ): Rf 0.41 (9:1 CHCl3:MeOH). Proton NMR (CDCl3): δ 7.9 (Ar-H), 7.41 (Ar-H), 7.06 (Ar-H), 6.90 (Ar-H), 5.29 (Ar-H), 3.64 (m, -O-CH-O-), 3.07 (s, 3H, NCH3), 1.86 (m, -O-CH2-CH2-); C-NMR NMR (CDCl3): δ 161.4, 155.1, 134.1, 127.7, 122.2, 118.0, 116.7, 89.3, 61.6, 31.0 (NCH3), 28.6, 27.6; MS (C11H13NO3) calcd. 207.090 found MH+208.3.
Conclusion
In this work different types of salicylate and salicylamide alcohols were synthesized for the preparation of phosphorodiamidates and ifosfamide pro drugs.
Acknowledgement
The author wish to thank Former Professor D. Farquar for his necessary guidance in the work and Dr. W. Bormmann, Former Professor, M.D. Anderson Cancer Center for his support and discussion in the work.
Conflicts of Interest
None
References
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.Nat Rev Drug Discov.,2008, 7(3), 255-70.
CrossRef - Wu, K. M. Pharmaceuticals (Basel)., 2009, 2(3), 77-81.
CrossRef - Buxton, I. L. O. Pharmacokinetics and Pharmacodynamics: The Dynamics of Drug Absorption, Distribution, Action, and Elimination. In: Brunton, L. L.; Lazo, J. S.; Parker, K. L. Goodman & Gilman’s the Pharmacological Basis of Therapeutics. 11th ed. New York: McGraw-Hill Medical Pub. Division,2005, 1-11.
- Goldstein, A.; Aronow, L.; Kalman, S. M. Principles of Drug Action: the basis of pharmacology. 2nd ed. NJ, USA: John Wiley & Sons; Hoboken,1974.
- Zawilska, J. B.; Wojcieszak, J.; Olejniczak, A. B. Pharmacol Rep.,2013, 65, 1-14.
CrossRef - Stella, V. J. J Pharm Sci.2010, 99, 4755-65.
CrossRef - Huttunen, K. M.; Raunio, H.; Rautio, J. Pharmacol Rev.,2011, 63, 750-71.
CrossRef - Rautio, J.; Meanwell, N. A.; Di, L. Nat Rev Drug Discov.,2018, 17(8), 559–587.
CrossRef - Rautio, J.; Kärkkäinen, J.; Sloan, K. B. Eur J Pharm Sci.,2017, 109, 146-161.
CrossRef - Najjar, A.; Najjar, A.; Karaman, R. Molecules.,2019, 16, 1-5.
CrossRef - Struck, R. F. Isophosphoramide mustard analogs and use thereof, PCT Int. Appl. 2000, 34 pp. CODEN: PIXXD2 WO. 2000071134 A1.
- Sladek, N. E. Pharmacol. Ther.,1988, 37, 301-355.
CrossRef - Sladek, N. E. Oxazaphosphorines, Metabolism and Action of Anticancer Drugs; Powis, G., Prough, R. A., Eds.; Taylor and Francis: London, 1987, 48-90.
- Friedman, O. M.; Myles, A.; Colvin, M Cyclophosphamide and Related phosphoramide Mustards, Advances in Cancer Chemotherapy; Rosowsky, A.; Marcel Dekker: Newyork, 1979, 143-204.
- Hill, D.L. A Review of Cyclophosphamide; Charles C. Thomas: Springfield, IL, 1975.
- Zon, G. Prog. Med. Chem.,1982, 19, 205-246.
CrossRef - Stec, W. J. Organophosphate Chem.,1982, 13, 145-174.
- Wang, Y. and Farquhar, D. J. Med. Chem.,1991, 34, 197-203.
CrossRef - Struck, R. F.; Kirk, M. C.; Witt, M. H.; Laster, W. R.Jr. Biomed. Mass Spectrom.,1975, 2, 46.
CrossRef - Struck, R. F.; Dykes, D. J.; Corbett, T. H.; Suling, W. J.; Trader, M. W. B. J. Cancer.,1983, 47, 15.
CrossRef - Friedman, O. M.; Wodinsky, I.; Myles, A. Cancer Treat. Rep. 1976, 60, 337.
CrossRef - Hipkings, J. H.; Struck, R. F.; Gurtoo, H. L. Cancer Res. 1981, 41, 3571.
- El-Din, N. S. Acta Pharm.,2000, 50, 239-248.
- Feldman, K. S.; Lawlor, M. D.; Sahasrabudhe, K. J. Org. Chem. 2000, 65, 8011-8019.
CrossRef - Zon, G.; Ludeman, S. M.; Brandt, J. A.; Boyd, V. L.; Ozkan, G.; Egan, W.; Shao, K. L. J. Med. Chem.,1984, 27, 466-485.
CrossRef - Brown, F. J. J. Med. Chem.,1989, 32, 807-826.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


