Solid Phase Synthesis of Biologically Active Benzimidazole Derivatives Catalysed by Methanesulphonic Acid-SiO2 under Solvent Free Condition
Arup Dattaa and Sanjay Roya*
and Sanjay Roya*
Department of Chemistry, Shibpur Dinobundhoo Institution (College), 412/1, G. T. Road (South), Howrah-711102, West Bengal, India
Corresponding Author E-mail: sanjayroyp@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/360325
Article Received on : 28 May 2020
Article Accepted on : 29 Jun 2020
Article Published : 29 Jun 2020
A simple an expeditious method was established for the synthesis of 2-Substituted-1H-Benzimidazole derivatives at 90º C using o-phenylenediamine and different aldehydes. It has been found that a mixture of Methanesulphonic acid-SiO2 to be an effective catalyst to generate moderate to high yield of a wide variety of 2-substituted benzimidazole derivatives through a simple workup process. Solid phase, rapid reaction, solvent free technique and simplicity of the reaction procedure are the advantages of this protocol.
Benzimidazole; Heterogeneous Catalyst; Solvent Free Condition; Solid Phase
Download this article as:| Copy the following to cite this article: Datta A, Roy S. Solid Phase Synthesis of Biologically Active Benzimidazole Derivatives Catalysed by Methanesulphonic Acid-SiO2 under Solvent Free Condition. Orient J Chem 2020;36(3). |
| Copy the following to cite this URL: Datta A, Roy S. Solid Phase Synthesis of Biologically Active Benzimidazole Derivatives Catalysed by Methanesulphonic Acid-SiO2 under Solvent Free Condition. Orient J Chem 2020;36(3). Available from: https://bit.ly/2ND39Fd |
Introduction
Environmentally benign chemical reactions have gained considerable interest in academic and industrial process for the past few years.1 Keeping in mind to protect our environment the use of hazardous, toxic, volatile organic solvents are reduced and solvent free technique2 is highly appreciated. Now a day’s organic reactions are carried out either by the use of phase transfer catalyst3a in aqueous medium3b, ionic liquids4 or solvent free technique.5 Since methanesulphonic acid in silica creates mild acidity under solvent free condition,6 so a library of compounds have been developed having neutral, acidic and base sensitive group in aldehydes. Benzimidazole moiety actually presents in naturally biological active substances such as vitamin B12 and purine bases. Benzimidazole exhibits various therapeutic purposes in medicinal chemistry7 like anticancer,8 antimicrobial,9 antifungal,10 antihypertensive,11 antiparasitic,12 antihistaminic,13 anti-inflmmatory,14 antiviral,15 activities. They also have different applications in fluorescence,16 crystal engineering,17 chemo sensing 18 and corrosion science.19 Therefore, synthesis of these compounds is very important to organic chemists and biologists. Over the few years these compounds have been synthesized by several synthetic process but two fundamental methods are generally utilized to develop the products. Nucleophilic attack of orthophenylenediamines with aromatic carboxylic acids and their derivatives such as nitrile, imides in strong acid20 like PPA, or in presence of catalyst at high-temperature or microwave irradiations21 have been applied to endorse the formation of benzimidazole. Second procedure was the reaction of orthophenylenediamine and aldehyde in presence of either mineral acids or transition metal salt in presence of oxidizing reagents.22 PEG-100 and 400 were also used under solvent free condition at high temperature.23 So from above discussion we can say most of the reported methods are inconsistent because of the following reasons; (a) use of toxic metallic compounds23(a) (b) applied high temperature above 1000C 23(b,d) (c) use of hazardous solvent23(c,e) (d) laborious workup procedure23(c) and (e) additive oxidizing reagents23(e) were used in the second step of the reactions. So a new approach is required to develop the benzimidazole in a very simple and as well as rapid process.
Results and Discussion
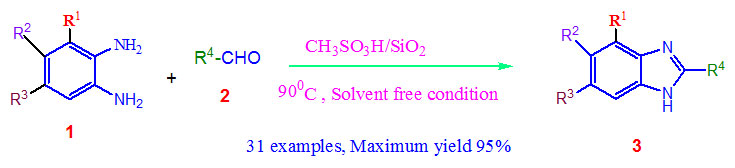
In modern organic synthesis, solvent-less condition is a very significant research area24(a) now a day’s. In continuation of the searching work for the creation of the modern methodology for benzimidazole production here we declare a one-pot, simple, rapid and easy workup method associated with the formation of benzimidazole derivatives using orthophenylenediamine with different aldehydes under solvent free condition at 900C in presence of organic methanesulphonic acid-Silica catalyst.
 |
Scheme 1: Synthesis of 2-substituted-1H-benzimidazoles catalysed |
Table 1: Study of the optimization of the reaction in different conditions between p-chlorobenzaldehyde(3.0 mmols) and o-phenylenediamine(3.2mmols) to the formation of benzimidazole (3f, Table 2):
|
Entry Number |
Methane sulphonic acid (mL) |
Silica (mg) |
Reaction Conditions Solvent (5 mL), Temperature |
Time (hrs) |
Yield (%) (Isolated) |
|
1 |
(-) |
(-) |
Free, Oil bath 90 oC |
15 |
45 |
|
2 |
0.1 |
A |
Free, Oil bath 50 oC |
15 |
45 |
|
3 |
0.2 |
A |
Free, Oil bath 50 oC |
10 |
50 |
|
4 |
0.5 |
A |
Free, Oil bath 90 oC |
8 |
60 |
|
5 |
0.6 |
A |
Free, Oil bath 90 oC |
8 |
75 |
|
6 |
0.8 |
A |
Free, Oil bath 90 oC |
8 |
80 |
|
7 |
1.0 |
A |
Free, Oil bath 90 oC |
6 |
95 |
|
8 |
1.5 |
A |
Free, Oil bath 90 oC |
6 |
95 |
|
9 |
2.0 |
A |
Free, Oil bath 90 oC |
6 |
95 |
|
10 |
1.0 |
B |
Free, Oil bath 90 oC |
6 |
95 |
|
11 |
1.0 |
A |
MeCN,Oil bath 90 oC, reflux |
15 |
75 |
|
12 |
1.0 |
A |
EtOH, Oil bath 90 oC, reflux |
15 |
78 |
|
13 |
1.0 |
A |
DMF, Oil bath 90 oC, reflux |
15 |
75 |
|
14 |
(-) |
(-) |
MeCN/ EtOH/ DMF, Oil bath 90 oC, reflux |
15 |
50 |
|
(+) means present and (–) means absent; A means Silica 0.5g and B means Silica 1g |
|||||
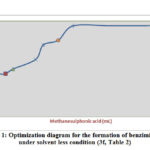 |
Figure 1: Optimization diagram for the formation of benzimidazole under solvent less condition(3f, Table 2) |
It has been observed that in presence of acid catalyst alone at appropriate temperature, condensation of the reaction leads to several by-products and consequently low yield of the products are obtained. Burning tendency of compound at 900C was reduced by the use of silica surface as a catalyst. Initial experiment was carried out on the study of catalytic activity of methanesulphonic acid-silica for the synthesis of benzimidazole (3f) by treating highly reactive substrate p-chlorobenzaldehyde (3 mmol) with o-phenylenediamine (3.2 mmol) in solvent free condition at 900C (Table 1). In absence of catalyst methanesulphonic acid reaction did not produce excellent yield of the product even after running for a long time of the reaction at 900C (Entry No 1, Table 1). The benzimidazole derivative formation increases with raising the volume of catalyst gradually at 900C and it was represented graphically (Fig 1) (Entry No 2-7, Table 1). But the best result was observed when methanesulphonic acid and silica were used 1 mL and 0.5g respectively and the reaction completed within 6 hrs monitoring by TLC measurement (Entry no 7, Table 1). No improvement of the yield of the product was determined even more than 1ml and 0.5g of the acid and silica were used (Entry No 8,9,10, Table 1) and not only that this method gave the poor result in presence of polar protic and aprotic solvents. (Entry No 11, 12, 13, 14, Table 1). It was also monitored that without silica low yield in the reaction was observed (No 1, 14, Table 1) and finally we have found the optimization of the reaction condition (Entry No 7, Table 1,) where both silica (0.5g) and methanesulphonic acid (1mL) are essential to carry out the reaction.
Table 2: Methanesulphonic acid-silica (1mL-0.5g) catalysed reactions of aldehydes with ortho phenylenediamines under solvent free condition at 900C
|
Entry number
|
Starting ortho– phenylenediamine 1 |
Product 3 |
Starting aldehyde 2 R4 |
Heating period (hours) |
Yield (%) (isolated) |
References |
||
|
R1 |
R2 |
R3 |
||||||
|
1 |
H |
H |
H |
a |
4-Me |
6 |
85 |
25(a,b) |
|
2 |
H |
H |
H |
b |
2-Me |
8 |
80 |
25(a,b) |
|
3 |
H |
H |
H |
c |
3-Me |
7 |
85 |
25(a,b) |
|
4 |
H |
H |
H |
d |
4-OH |
8 |
90 |
25(c) |
|
5 |
H |
H |
H |
e |
2-OH |
9 |
85 |
25(d) |
|
6 |
H |
H |
H |
f |
4-Cl |
5 |
95 |
23(b) |
|
7 |
H |
H |
H |
g |
3-Cl |
5 |
85 |
23(b) |
|
8 |
H |
H |
H |
h |
4-Br |
4 |
92 |
23(d) |
|
9 |
H |
H |
H |
i |
2-OMe |
6 |
90 |
23(c) |
|
10 |
H |
H |
H |
j |
4-OMe |
7 |
85 |
26 (c) |
|
11 |
H |
H |
H |
k |
4-NO2 |
4 |
90 |
27(a) |
|
12 |
H |
H |
H |
l |
2-CN |
8 |
85 |
25(a) |
|
13 |
H |
H |
H |
m |
4-CN |
6 |
88 |
25(a) |
|
14 |
H |
H |
H |
n |
R4-C6H4 =2-furanyl |
7 |
85 |
27(a) |
|
15 |
H |
H |
H |
o |
Ph |
5 |
88 |
27(a,b) |
|
16 |
H |
Cl |
Cl |
p |
4-NO2 |
4 |
92 |
23(b) |
|
17 |
H |
Cl |
Cl |
q |
4-CN |
4 |
85 |
27(b) |
|
18 |
H |
Me |
H |
r |
3-OH |
4 |
92 |
26(b) |
|
19 |
H |
Me |
H |
s |
2-NO2 |
4 |
88 |
26(c) |
|
20 |
H |
Me |
H |
t |
NMe2 |
8 |
80 |
26(c) |
|
21 |
H |
Me |
H |
u |
2-Cl |
6 |
82 |
26(c) |
|
22 |
H |
Me |
Me |
v |
4-Cl |
5 |
92 |
23(b) |
|
23 |
H |
Me |
Me |
w |
4-OMe |
8 |
85 |
23(b) |
|
24 |
H |
Me |
Me |
x |
4-CN |
5 |
88 |
23(b) |
|
25 |
H |
Me |
Me |
y |
3-NO2 |
4 |
90 |
23(b) |
|
26 |
H |
Me |
Me |
z |
2,5-(OMe)2 |
7 |
82 |
23(b) |
|
27 |
H |
Me |
Me |
a’ |
Me |
7 |
75 |
23 |
|
28 |
H |
H |
H |
b’ |
Me |
7 |
78 |
23 |
After the success of optimization method then we successfully applied this optimized condition for a variety of aromatic and aliphatic consisting of different electronic behavior groups for product development and the isolated yield of the products were up to 75-95% (Table 2). In this methodology four different ortho-phenylenediamines were used under solvent free condition and reaction proceeds smoothly in all cases. Electron withdrawing group (like NO2, CN) gave better yield but electron donating group (like OMe, NMe2) gave comparatively low yield of the products. Substitution of the same group at different position in aromatic ring also gave different yield because of electronic and steric factor. The mechanism of the reaction has been proved in several published papers24(b). This reaction was monitored in argon chamber but intermediate addition product imines were confirmed by checking the TLC experiment and that’s why overall experiment was done in open atmosphere. After the success of the above reaction then we tried to apply this condition for the formation of bis benzimidazole. At 90oC temperature incomplete conversion was observed in several cases and to complete the reaction long time was required. It was found that if the temperature can be increased then this reaction was complete in less time and our goal was to do it in less time. So this condition was slightly modified and observed that at 120oC the good yield was obtained and other conditions were remaining unchanged. In connection of the reaction two different dialdehydes and two different orthophenylenediamine were used and yield shown in the Table 3.
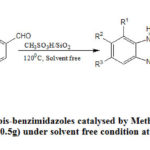 |
Scheme 2: Synthesis of bis-benzimidazoles catalysed by Methanesulphonic acid-silica |
Table 3: Methanesulphonic acid-silica catalysed reactions of bis-aldehydes with ortho phenylenediamines under solvent free condition at 1200C.
|
Entry Number |
Starting ortho– phenylenediamine 1 |
Product 3 |
Starting aldehyde 2 |
Heating period (hours) |
Yield (%) (isolated) |
References |
||
|
R1 |
R2 |
R3 |
||||||
|
1 |
H |
H |
H |
c’ |
1,4-dialdehyde |
8 |
83 |
23 |
|
2 |
H |
H |
H |
d’ |
1,3-dialdehyde |
9 |
79 |
23 |
|
3 |
H |
Cl |
Cl |
e’ |
1,3-dialdehyde |
9 |
76 |
23 |
The following advantages of this method came out from the above discussion and they are as follows (a) The reaction is environmentally benign because no hazardous solvent is used in the reaction; (b) Medium temperature was used to complete the reaction compare to other published papers23(b); (c) No additive oxidizing reagent is required in the second step for oxidation; (d) Catalytic amount methanesulphonic acid in 0.5g Silica is sufficient in this reaction smoothly; (e) Silica is non-toxic, inexpensive and non-deformable compound24(c) and it was reused for three times in the reactions without loss of its efficacy. So recycle property of the catalyst was explored in this methods as shown in Figure 2; (f) Since the compound was solid so ethanol was used to crystallization directly after separation of silica by simple filtration technique that means no need of hectic column chromatography; (g) This method can be used to make compounds in large scale.
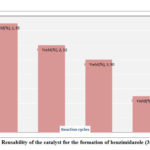 |
Figure 2: Reusability of the catalyst for the formation of benzimidazole (3f, Table 2) |
Experimental
All chemicals and solvents are used AR grade. All aromatic aldehydes, o-phenylenediamine, methane sulphonic acid and silica were purchased from Spectrochem, Pvt. Ltd. Mumbai, India. 1H and 13C NMR spectra were recorded on Bruker 300 MHz spectrometer at room temperature CDCl3 and DMSO-d6 solvents were purchased from Spectrochem. FT-IR spectra were recorded on a Perkin-Elmer spectrometer. Organic solvent was dried with Magnesium sulfate. Melting points were measured with an electrical apparatus. Silica coated TLC plate was used to monitor the reaction.
General Procedure
A mixture of 4-Chlorobenzaldehyde (420mg, 3mmol, 2) and orthophenylenediamine (346mg, 3.2mmol, 1) were mixed with methanesulphonic acid(1mL) and silica (0.5g) in a 50 mL round-bottomed flask and it was heated in an oil bath at 900C. Time to time the reaction was observed by silica coated TLC plate in presence of ethyl acetate and petroleum ether as an eluent. The reaction completed after the formation of brown spot within 4-9 hrs in iodine chamber. Then the crude product was cooled and diluted with ethylacetate, and filtered. The filtrate was washed three times with 50 mL saturated aqueous bicarbonate solution. It was washed three times with 30mL brine solution. Then it was concentrated in a rotary evaporator under vacuum at room temperature. The product was crystallized directly from hot aqueous ethanol to produce the pure products. For bis benzimidazole same method was used but change of temperature was 1200C. Character analysis of the product was compared by FT-IR, 1H NMR and 13C NMR data with a good quality of known compounds.
Selected Characterization Data for Synthesized Compounds
4-(5,6-Dichloro-1H-Benzimidazol-2-yl)Benzonitrile (3q)
Characteristic: Light yellow solid.M.P. >320oC. (Aq-EtOH), FT-IR (cm-1): 3294, 2232, 1610, 1440, 1414, 1291, 1100 and 846.
1H NMR (300 MHz, DMSO-d6): d 12.91 (brs, 1H, N-H), 8.30 (d, J = 8.4 Hz, 2H, C3 and C5 proton), 7.90–7.73 (brs C4, C2 and C6 proton), 7.61 (brs, 1H, C7 proton). 13C NMR (75MHz, DMSO-d6): d 150.63, 132.51, 13 1.28, 126.15, 117.04, 111.79. Analytical Calculation for C14H7N3Cl2 (%): C 58.36, H 2.45, N 14.59; Found: C 58.41, H 2.30, N 14.52.
2-(4-Chlorophenyl)-5,6-Dimethyl-1H-Benzoimidazole (3v)
Characteristic: Light Grey solid. M.P. : 260 oC (Aq-EtOH). FT-IR (cm-1): 2925, 2374, 1593, 1437, 1092, 1011, 834, 725 and 534.
1H NMR (300 MHz, DMSO-d6): d 2.29 (s, 6H, Me proton), 7.24 (1H, s, C4 proton), 7.37 (s, 1H, C6 proton ), 7.51 (dd, 2H, ortho to Cl), 8.13 (d, 2H, meta to Cl), 12.63 (s,1H, N-H). 13C NMR (75 MHz, DMSO-d6): d 130.21, 131.54, 133.62, 134.11, 142.52, 149.31. Analytical Calculation for C15H13ClN2 (%): C: 70.18, H: 5.10, N: 10.91; Found: C: 70.09, H: 5.03, N: 10.81.
4-(5,6-Dimethyl-1H-Benzimidazol-2-yl)-Benzonitrile (3x)
Characteristic: Light yellow solid. M.P. : 199 oC. (Aq-EtOH). FT-IR (cm-1): 3423, 3067, 2925, 2370, 2225, 1610, 1444, 1281 and 1000.
1H NMR (300 MHz, CDCl3): d 2.36 (m, 6H,2Me), 7.38 (s, 2H, C4 and C6 proton), 7.79 (d, 2H, meta to CN ), 8.30 (d, 2H, Ortho to CN). 13C NMR (75 MHz, CDCl3):d 18.2, 109.7, 116.7, 124.8, 129.6, 130.8, 132.6.
Analytical Calculation for C16H13N3 (%): C: 77.71; H: 5.30; N: 16.99 Found: C: 77.61; H: 5.24, N: 16.80
2-(2′, 5′-Dimethoxyphenyl)-5,6-Dimethyl-1H-Benzimidazole (3z)
Characteristic: Light white solid.mp:270 oC. (Aq-EtOH);FT–IR (cm-1): 2925, 2375, 1523, 1345, 1129, 851 and 698.
1H NMR (300 MHz, CDCl3): d 11.85(s, 1H, N-H), 7.81(d, 1H, C6′ proton), 7.38(s, 1H, C4 proton), 7.33(s, 1H, C7 proton), 7.11(d,1H, C4′ proton), 6.98(d, 1H, C3′ proton), 3.92(s, 3H, C2′-OMe), 3.75( s, 3H, C5′-OMe), 2.29 and 2.27 (s, 6H, 2Me). 13C NMR (75 MHz, CDCl3): d 153.27, 150.96, 147.93, 141.42, 133.38, 130.77, 129.92, 118.90, 118.57, 116.75, 113.54, 113.44, 112.08, 56.17, 55.59, 20.25, 20.09. Analytical Calculation for C17H18N2O2 (%): C 72.32, H 6.43, N 9.92; Found: C 72.24, H 6.51, N 9.81
5,6-Dichloro-2-[3′-(5,6-Dichloro-1H-Benzimidazol-2-yl)Phenyl]-1H-Benzimidazole (3e’)
Characteristic: Pale yellow solid; M.P: >320 °C (EtOH); FT-IR (cm-1): 3409, 2925, 2856, 2197,1448, 1298, 1098 and 864.
1H NMR (300 MHz, DMSO-d6) δ: 13.43 (brs, 2H, 2N-H), 8.96 (s, 1H, C2′H proton), 8.22 (d, 2H, C4′ and C6′ protons), 8.00–7.62 (m, 5H, C4, C7 and C5′ protons); 13C NMR (75 MHz, DMSO-d6) δ: 153.2, 143.4, 134.4, 130.2, 129.9, 128.4, 125.3, 125.0, 124.6, 120.1, 113.0. Analytical Calculation for C20H10N4Cl4; C: 57.18, H: 2.40, N: 6.67%. Found: C: 57.30, H: 2.34, N: 6.50%.
Conclusion
We have developed a simple green methodology for the preparation of a series of benzimidazole and few bis benzimidazoles derivatives under solvent free condition in presence of heterogeneous catalyst. Simple, mild, rapid reaction, easy workup procedure and excellent yield are the main advantages of this protocol. Not only that recoverable silica is reused for more than three times which is another incidental advantage of this reaction. So in near future this method may be associated with compliance with the Eco-friendly procedure.
Acknowledgement
AD andSR are thankful to the Department of chemistry, shibpur Dinobundhoo Institution (College) for laboratory and computational facilities.
References
- Anastas PT.; Warner JC.; Green Chemistry, Theory and Practice; Oxford University Press: Oxford. 1998; (b) Clark J.; Macquarrie DM.; Handbook of Green Chemistry; Blackwell: Oxford. 2002.
- Sharma H.; Singh N.; Jang DO.; Green Chem. 2014, 16, 4922-4930.
- Vasudevan VN.; Rajender SV.; Green Chem. 2001, 3, 146. (b) Grieco PA.; Organic Synthesis in Water; Blackie Academic and Professional: London,
- Mikami K.; Green Reaction Media for Organic Synthesis, Wiley-VCH, 2003.
- Tanaka K.; Solvent-Free Organic Synthesis; Wiley-VCH, 2003.
- Sharghi; Asemani O.; Synthetic Commun, 2009, 39, 860–867.
- Arienti KL.; Brunmark A.; Axe FU.; McClure K.; Lee A.; Blevitt J.; Neff DK.; Huang L.; Crawford S.; Pandit CR.; Karlsson L.; Breitenbucher JG.; Med. Chem. 2005, 48(6),1873-1885.
- Soni B.; Ranawat MS.; Bhandari A.; Sharma R.; International Journal of Drug Research and Technology, 2012, 2, 479-485.
- Elnima EI.; Zubair MU.; Al-Badr AA.; Antimicrob Agents Chemother. 1981, 19(1), 29-32. (b) Kazimierczuk Z.; Upcroft AJ.; Gorska A.; Starooeciak B.; Laudy A.; Acta Biochim. Pol. 2002, 49(1), 185-195.
- Maxwell WA.; Brody G.; Environ. Microbiol. 1971, 21(5), 944-945. (b) Kılcıgil AG.; Altanlar N.; Turk. J. Chem. 2006, 30, 223-228.
- Rithe SR.; Jagtap RS.; Ubarhande SS.; Rasayan J Chem, 2015, 8(2), 213-217. (b) Shah D I.; Sharma M.; Bansal V.; Bansal G.; Singh M.; J. Med. Chem, 2008, 43,1808-1812.
- Navarrete VG.; Cedillo R.; Hernández CA.; Yépez L.; Hernández LF.; Valdez J.; Morales R.; Cortés R.; Hernández M.; Castillo R..; Med. Chem. Lett. 2001, 11, 187-190.
- Terzioğlu N.; van Rijn RM.; Bakker, RA.; De IJ.; Esch, P.; Leurs R.; Med. Chem. Lett., 2004, 14, 5251-60.
- Chen G.; Liu Z.; Zhang Y.; Shan X.; Jiang L.; Zhao Y.; He W.; Feng Z.; Yang S.; Liang G.; ACS Med. Chem. Lett. 2013, 4, 69-74.
- Cheng J.; Xie J.; Luo X.; Bioorg Med Chem Lett., 2005, 15, 267-269.
- Choudhury P.; Panja S.; Chatterjee A.; Bhattacharya P.; Chakravorti S.; Photochem. Photobiol. A 2005, 173, 106-113.
- Matthews CJ.; Broughton V.; Bernardinelli G.; Melich X.; Brand G.; Willis AC.; Williams AF.; New J. Chem. 2003, 27, 354-358.
- Wong WWH.; Vickers MS.; Cowley AR.; Paul RL.; Beer PD.; Biomol. Chem. 2005, 3, 4201-4208. (b) Singh N.; Jang DO.; Org. Lett. 2007, 9, 1991-1994.
- Khaled KF.; Acta. 2003, 48, 2493-2503. (b) Roque JM.; Pandiyan T.; Cruz J.; Garcı´a-Ochoa E.; Corros. Sci. 2008, 50, 614-624.
- Grimmet MR.; Academic Press: San Diego, 1997.
- (a) Lu J.; Yang B.; Bai Y.; Synth. Commun. 2002, 32, 3703-3709. (b) Wang Y.; Sarris K.; Sauer DR.; Djuric SW.; Tetrahedron Lett. 2006, 47(28), 4823-4826.
- Bahrami K.; Khodaei MM.; Kavianinia I.; 2007, 4, 547-550.
- Ke F.; Zhang P.; Xu Y.;Lin X.; Lin J.; Lin C.;Xu J.; Synlett, 2018,29, 2722-2726. (b) Mukhopadhyay C.; Tapaswi PK.; Tetrahedron Lett. 2008, 49, 6237-6240. (c) Du LH.; Wang YG.; Synthesis. 2007, 5, 675-678. (d) Hubbard JW.; Piegols AM.;. S€oderberg BCG.; Tetrahedron, 2007, 63, 7077–7085. (e) Birajdar SS.; Hatnapure DG.; Keche PA.; Kamble MV.; Research Journal of Pharmaceutical, Biological and Chemical Sciences, 2014, 5(1), 487-493.
- Kamal A.; Reddy DR.; Rajendar; Tetrahedron Lett. 2006, 47(13), 2261-2264. (b) Mukhopadhyay C.; Tapaswi PK.; Butcher.; RJ.; J. Chem. 2009, 62, 140–144. (c) Banerjee AK.; Mimo’ MSL.; Vegas WJV.; Russian Chemical Reviews, 2001, 70 (11), 971 – 990.
- Shen M.; Driver TG.; Lett. 2008, 10, 3367–3370. (b) Eren B.; Bekdemir Y.; Química Nova, 2014, 37(4), 643-647. (c) Chakrabarty M.; Karmakar S.; Mukherjee A.; Arima S.; Harigaya Y.; Heterocycles, 2006, 68, 967. (d) Ma H.; Han X.; Wang Y.; Wang J.; Heterocycles 2007, 71, 1821.
- Du HL.; Wang GL.; Synthesis, 2007, 5, 675-678. (b) Mukhopadhyay C.; Tapaswi PK.; Butcher RJ.; Australian journal of chemistry, 2009, 62(2).140-144.
- Das, H. Holla, Y. Srinivas, Tetrahedron Lett. 2007, 48, 61-64. (b) Mukhopadhyay C.; Tapaswi PK.; Catalysis Communications, 2008, 9, 2392–2394.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


