The Potential use of Geraniol Esters from Citronella Oil as Anticancer Agents
Galuh Widiyarti*, Megawati Megawati and Muhammad Hanafi
Research Center for Chemistry, Indonesian Institute of Sciences (LIPI) Kawasan PUSPIPTEK, Serpong, South Tangerang City, Banten 15314, Indonesia.
Corresponding Author E-mail: galuh.laksmono@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/350310
Article Received on : 26-02-2019
Article Accepted on : 02-05-2019
Article Published : 07 May 2019
Geraniol which is mainly contained in citronella oil is one of the Indonesian natural products with anticancer potential. In this study, synthesis of geranyl butyrate, geranyl caproate, and geranyl caprylate from geraniol was conducted using sodium hydroxide as a catalyst. The aim was to enhance the anticancer activity of geraniol as a starting material. In order to achieve this, its esters were identified through the use of Gas Chromatography-Mass Spectroscopy (GCMS), Fourier Transform Infra Red (FTIR), Thin Layer Chromatography (TLC), and Proton Nuclear Magnetic Resonance (1H-NMR). They were analyzed for their potential as anticancer agents through Brine Shrimp Lethality Test (BSLT) against Artemia salina Leach, Mosmann method against murine leukemia (P388) cells and normal (Vero) cells. It was found that geraniol esters have the potential to be anticancer compounds. This was indicated by LC50 values of 0.96-1.46 µg/ml against A. salina L, IC50 values of 22.34-32.29 µg/ml against P388 cells, and very less cytotoxic effect on Vero cells with IC50 values of 116.08-172.93 µg/ml. Therefore, there is an expectation that acyclic ester compounds should be used in treating cancer.
KEYWORDS:Anticancer; BSLT; Citronella Oil; Ester; Geraniol; Mosmann
Download this article as:| Copy the following to cite this article: Widiyarti G, Megawati M, Hanafi M. The Potential use of Geraniol Esters from Citronella Oil as Anticancer Agents. Orient J Chem 2019;35(3). |
| Copy the following to cite this URL: Widiyarti G, Megawati M, Hanafi M. The Potential use of Geraniol Esters from Citronella Oil as Anticancer Agents. Orient J Chem 2019;35(3). Available from: https://bit.ly/2WuvyQG |
Introduction
Cancer has been discovered to be one of the major reasons for human death through the globe and, in Indonesia, it is ranked second after heart disease with its patients increasing on a yearly basis.1-3 However, it can be treated by surgery, radiation, chemotherapy, and through drugs but no satisfactory result has been obtained from any of these methods. Therefore, there is a need to conduct research on the use of bioactive or synthesizing analog compounds in treating cancer. It is expected to provide a drug with lower toxicity, higher activity, more selectivity.
In recent years, there have been extraction of different chemo-preventive and chemotherapeutic compounds applied in the prevention of cancer from some plants because of their usefulness in researching new bioactive compounds.4-7 In Indonesia, Citronella is an example of these compounds and it is derived from Lemongrass plant, a genus of Cymbopogon and family of Poaceae. Different kinds of essential oils mostly utilized in cosmetics, perfume, sanitary, food, and pharmaceuticals industries throughout the world are made from it. It is, however, further divided into three based on source, namely Ceylon derived from Cymbopogon nardus Lenabatu W. Wats.; Java from Cymbopogon winterianus Jowit; and Lemongrass oil derived from Cymbapogon citrates DC Stapt.8 Indonesia is the third largest producer of the oil in the world, after China and Vietnam9 and almost all their products are dominated by the Java type which are traditionally used asmedicine, repellent, toothache and body warmers.
Citronella oil and its compounds have been observed to have the ability to be applied in bioactivity processes as antimicrobial, antibacterial, antifungal, antiviral, antiparasitic,10 antioxidant and anti-inflammatory agents.11-13 They mostly contain monoterpene compounds such as citronellol, citronellal, and geraniol14 which have been reported to be effective against cancerous cell proliferation and inhibition of DNA synthesis. This shows that they are one of the new classes of cancer chemo-preventive compounds.13,15,17 Most of the raw materials used in the production of synthetic drugs in Indonesia are gotten from other countries, therefore, local natural materials like geraniol mostly found in citronella oil are need to be utilized.
Fatty acids such as butyric (C4), caproic (C6), and caprylic (C8) acids have also been reported to function in bioactivity as antiviral, antifungal, antimicrobial, and potentially as antineoplastic agents.18,19 Hence, the combination of the ester compounds derived from geraniol with these fatty acids could be used in the chemoprevention of cancer. Furthermore, combining caproate synthesized from citronellol and caproic acid in palm kernel oil has also been reported to be effective in inhibiting murine leukemia (P388) cells growth through the increment of its cytotoxic activity. The 38.49 and 10.63 µg/ml concentration have also been found to inhibit 50% of murine leukemia (P388) cells growth (IC50).20
Therefore, this study was aimed at synthesizing geranyl butyrate, geranyl caproate, and geranyl caprylate using sodium hydroxide (NaOH) as a base catalyst to enhance the anticancer activity of geraniol derivatives. The geraniol esters were identified by using TLC, FTIR, GCMS, and 1H-NMR. Their potency as chemoprevention agents for cancer was tested against nauplii of A. Salina using BSLT method. This is generally considered as a bench top pre-assay to discover the cytotoxic agent for the anticancer compound.21,22 The MTT method was also used to check their cytotoxic activity against murine leukemia (P388) cells.23-24 The same method was also used to test them against normal (Vero) cells.25
Materials and Methods
The starting materials include geraniol from Aroma Essence Prima with 90% purity, butyric (Sigma Aldrich B1754), caproic, and caprylic acids from Sumi Asih with 99% purity. The catalyst used was NaOH (E.Merck 106469) with 1% of HCl solution as a netraulizer. The standard procedure was used in drying and distilling all the solvents including n-hexane, ethyl acetate (EtOAc), and water. Furthermore, mass and molecular structure spectroscopy, in vitro cytotoxic activity and BSLT test were conducted using analytical grade chemicals.
General Procedure
The precoated silica gel plates were analyzed through the use of the TLC (Kiesel gel 60F254 0.25 mm), and there was visualization of the spots in them under the UV light (254 and 365 nm) irradiation, sprinkled with 10% sulphuric acid solution under 110°C. A Merck 64271 (70-230 mesh) was used to conduct column chromatography on the silica gel. The measurement of IR spectra was done on an FTIR Shimadzu prestige 21 with the use of KBr pellets while the evaluation of mass spectrum (MS) was conducted through the use of GC Agilent Technologies 7890B with 5977A MSD. NMR Jeol spectrophotometer was used to record 1H-NMR spectrum for 500 MHz in deuterio chloroform. It is important to point out that the chemical shifts (δ) were in ppm and downfield from CDCl3 at δ 7.26 ppm.
Synthesis of Geraniol Ester Compound
10 mmol of geraniol, 12 mmol of butyric acid (or caproic acid or caprylic acid), and 5% (w/w) of NaOH were arranged in a 250 mL round-bottomed flask. The mixture was stirred and heated at 80°C for 8 hours. This was followed by the neutralization of the reaction by 1% of HCl solution for the purpose of separating it from the NaOH catalyst. EtOAc was used to extract the reaction mixture and the organic phase was washed through the use of distilled water, dried over anhydrous Na2SO4, and concentrated under reduced pressure at 45°C under vacuum to produce the crude ester product. The analysis of the products was conducted by TLC through the use of eluent of n-hexane: EtOAc = 95:5 and further purification was conducted through the use of column chromatography eluted with n-hexane, a gradient of EtOAc to 100%. The identification of pure ester product was conducted through the application of spectroscopic methods (MS, IR, 1H-NMR).
BSLT Test
Hatching of the brine shrimp was the first thing to be conducted. This was done by placing about 50-100 mg brine shrimp eggs (A. salina L) in a vessel under light regime conditions and through the application of artificial seawater from commercial sea salt, they were hatched and grew to larvae with 48 hours of incubation under room temperature (25-29°C). There was collection of the larvae (nauplii) through the use of pipette leaving their shells behind.
The application of the Meyer method to conduct the BSLT analysis was the second step.21,22 To do this, there was dissolution of four (4) mg of the water-soluble samples in artificial seawater to produce 2 mg/ml solution while the water-insoluble compounds were dissolved in DMSO. The preparation of 10, 100, 500, and 1000 μg/ml of the solution followed this and there was the transfer of about 100 μl of seawater with 10-11 shrimp larvae of A. salina to the 96-well microplates. The DMSO groups were used as the control of the experiment while there was addition of approximately 100 μl of each sample to the test wells and each of the concentrations was made to be in three replicates. There was incubation of the solutions for a period of 24 hours at room temperature under illumination after which the live and dead larvae present in every well were counted. The determination of the 50% lethal concentration of the sample solutions (LC50) was conducted through the use of Probit analysis.
In vitro Anticancer Analysis Against P388 and Vero Cells using the MTT Method
Cytotoxicity test against murine leukemia (P388) cells was conducted by using Mosmann method through the application of the MTT reagent.23 This was established on the reduction of yellow water-soluble substrate 3- 4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) into an insoluble blue colored formazan product by the mitochondria succinate dehydrogenase enzymes in living cells. The measurement of the activity level was conducted by using the cells viability because of the fact that MTT can only be reduced in cells that are metabolically active.26
The first step of the process was growing the cell cultures. This was conducted by incubating the cells in RPMI 1640- fetal bovine serum (FBS) at 37°C with a flow of 5% of CO2 for 24 hours and observed under an inverted microscope. The attachment of the cells to the walls of flasks caused the replacement of the medium with new serum, after which they were primarily incubated before the confluent growth was reached. This refurbishment was continuously conducted until the sufficient number of cells needed for testing was obtained.
The second step was the preparation of cells for testing. The murine leukemia (P338) cells line were provided from Meijo Seika Co., Ltd., Tokyo Japan, while kidney of an African green monkey (Vero, ATCC CCL-81) with normal cells was obtained from the Department of Biology Universit as, Indonesia. Each cell line was cultured in a suitable medium to obtain the desired growth and their growth curve was plotted. The medium was discarded, washed, and added to the RPMI medium after the adequate number of cells needed for the test has been. There was transfer of the cells into sterile tubes where they were centrifugated for 5 minutes at 1200 rpm. The supernatant was cast aside and the sediment of the cell was put into the RPMI 1640 medium containing 10% FBS. The suspension of the cell was taken and the calculation of their density was conducted through the use of hemocytometer under a microscope. The formula below was used in calculating the number of cells:
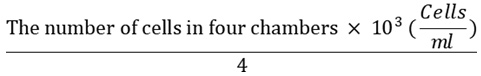
The calculated figure was used to dilute the cells with medium RPMI-serum to obtain 2 X 104 cells/ml of cell suspension.
The third step of the process was the cytotoxicity test. This was conducted by disturbing the cells with densities 1-2 X 104 cells/well in RPMI 1640 culture medium in 96 well-plates and incubating them in a 5% of CO2 incubator at 37°C for 24 hours and later stocked at the bottom wells. The next day, the media was taken, washed with FBS, and 100 μl of culture medium containing DMSO or sample with any of 100, 50, 25, 10, 5, 2.5 and 1 μg/ml concentrations were added and incubated for 48 hours. The sample was dissolved in DMSO and diluted using phosphoric acid buffer solution (PBS). Culture medium was then removed, washed with 100 μl of FBS, and an addition of 100 μl of a new culture medium containing MTT 5 mg/ml was made. It was incubated at 37°C in 5% of CO2 for 4 hours. Since it has been established that living cells will interact with MTT to form purple formazan crystals. The medium containing MTT was removed after 4 hours, it washed with PBS, and 200 μl of isopropanoate solution was added and incubated at room temperature for 12 hours to completely dissolve the formazan crystals. The bio-reduction of MTT was assessed by measuring the absorbance of each well at 550 nm through the use of ELISA reader. The regression equation and its IC50 (i.e. the concentration that caused the death of 50% of the population of the cells) were determined. The cytotoxicity was conducted in three replicates. It is important to point out that the IC50 value determines how effective a compound can be in suppressing the biological function of a sample that is still active. The calculation of this value is conducted by using the linear regression analysis.23-26
Results and Discussion
As stated earlier, the synthesis of geraniol esters from geraniol with butyric acid, caproic acid, and caprylic acid was conducted by using a base catalyst (NaOH) at 80°C for 8 hours as shown in Figure 1.
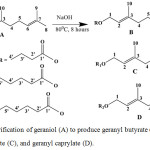 |
Figure 1: Esterification of geraniol (A) to produce geranyl butyrate (B), geranyl caproate (C), and geranyl caprylate (D). |
Oily liquids were obtained as the products and the analysis of the TLC revealed that there are ester compounds in them as represented by a spot with Rf value of 0.4, 0.4, and 0.3 for geranyl butyrate, geranyl caproate, and geranyl caprylate compounds, respectively. These spots were lower than the 0.8 Rf value of geraniol when used as a starting material. The crudes were further purified by using chromatography column using silica gel as the stationary phase and a solvent mixing ratio for n-hexane and gradient of EtOAc at 100% as the mobile phase. It obtained using a ratio 98:2, while pure geranyl caprylate compound was obtained at 95:5. The percentage yield of pure geranyl butyrate, geranyl caproate, and geranyl caprylate were 66.94% (0.75g), 76.72% (1.94g) and 54.92% (1.54g), respectively.
The formation of a new group of geranyl butyrate with 1722.43 cm-1 wave number (Vmax) was showed by IR spectrum. This stretched the vibration of carbonyl (C=O) of the ester formed with a variation from the absorption of carbonyl (C=O) of butyric acid at 1707 cm-1 wave number (V max). Furthermore, the formation of a new group of geranyl caproate with 1718.93 cm-1 was displayed by the IR spectrum. This stretched the vibration of carbonyl (C=O) of the ester formed with a variation from the absorption of carbonyl (C=O) of caproic acid at 1705.7 cm-1. The formation of a new group of geranyl caprylate with 1718.53 cm-1 was displayed by the IR spectrum. This stretched the vibration of carbonyl (C=O) of the ester formed with a variation from the absorption of carbonyl (C=O) of caprylic acid at 1702 cm-1. These spectra can be seen in Figure 2.
 |
Figure 2: IR spectra of geranyl butyrate (A), geranyl caproate (B), and geranyl caprylate (C). |
The MS of geraniol esters was identified using GCMS. For the geranyl butyrate, it showed a dominant peak at 14.61 minutes of retention time corresponding to a molecular formula of C14H24O2 of geranyl butyrate (the IUPAC name is butyric acid 2,2-dimethyl-3,7-dimethyl-2,6-octadienyl ester) at MS equals to 224.34 g/mol. For both geranyl caproate and geranyl caprylate, it was found that the MS of geranyl caproate (hexanoic acid 3,7-dimethyl-6-octenyl ester with molecular formula of C16H28O2) and geranyl caprylate (octanoic acid 3,7-dimethyl-2,6-octadienyl ester with molecular formula of C18H32O2) were 252.39 and 280.45 g/mol with the indication of dominant peaks at retention times of 17.30 and 19.39 minutes, respectively.
The GCMS chromatograms also showed another peak at retention times of 10.92, 11.33 and 11.21 minutes for geranyl butyrate, geranyl caproate and geranyl caprylate, respectively. This can be attached to the unreacted reactant residuals of the geraniol. This shows that the esterification reaction is not perfect yet, therefore, there was a need for further optimization.
Figure 1 and Table 1 show the 1H-NMR spectra data for both geraniol and geraniol ester compounds with respect to the molecular structure they possess. It reveals that new chemical shifts are formed by methyl (CH3), methylene (CH2) and methyne (CH) groups of carboxylic acid or fatty acids and another shift at δH = 4.13-4.59 ppm (2H, t, 7.2-7.4 Hz). This involved 2 protons (H) of the methylene (CH2) group used in forming triplet adjacent to CH2 group and downfield due to its interaction with the electronegativity of the oxygen and carbonyl (C=O) of the ester formed. These are not the same with that of 2 protons (2H) of CH2 group used in forming triplet as adjacent to CH group and downfield due to its interaction with the electronegativity of alcohol oxygen (OH) in the geraniol at δH =3.65 ppm (2H, t,) as it can be seen in Table 1.
The data for geranyl butyrate obtained from the 1H-NMR spectrum showed that at δH = 4.56 ppm (2H, d, 7.2 Hz), a chemical shift occurred for 2 protons (H) of the CH2 group used in forming doublet as adjacent to CH group and downfield due to its interaction with the electronegativity of oxygen and ester carbonyl (C=O) group. Another shift was experienced at δH = 5.41 and 5.31 ppm (1H, t, 7.2 Hz) for one proton of 2 CH groups interacting with 2 protons from the CH2 group, and downfield because of its double bond. A chemical movement was observed at δH = 2.32 and 2.16 ppm 2(2H, t, 7.2 Hz) for 2 protons of 2 CH2 groups interacting with CH2 group. Another shift was seen at δH = 1.51 and 1.67 (3H, s) for 3 CH3 groups interacting with the quaternary carbon atoms (C). At δH = 1.79 (2H, m, 7.5 Hz), chemical shifts of 2 protons of CH2 group used in forming multiplet were observed due to its interaction with 3 protons of the CH3 group and 2 protons of the CH2 group while another was seen at δH = 2.16 (2H, m, 7.2 Hz) for 2 protons of CH2 group used to form quartet shifted due to its interaction with 2 protons of the CH2 group and 1 proton of the CH group, and at δH = 0.9 ppm (3H, t, 7.8 Hz), 3 protons of CH3 group interacting with CH2 group shifted.
The data for geranyl caproate from the 1H-NMR spectrum revealed that there was a chemical shift at δH = 4.13 ppm (2H, d, 7.2 Hz) for 2 protons (H) of the methylene (CH2) group that formed doublet as adjacent to CH group and downfield because of its interaction with the electronegative oxygen and ester carbonyl (C=O) group. Another was observed at δH = 5.09 and 5.39 ppm (1H, t, 7.3 Hz) for one proton of 2 CH groups interacting with 2 protons from the CH2 group, and downfield because of its double bond. The was observable chemical shift at δH = 2.01 and 2.06 ppm (2H, t, 7.2 Hz), for 2 protons of 2 CH2 groups that interacted with CH2 group and at δH = 1.67 and 1.58 ppm (3H, s), 2 CH3 groups that interacted with 2 protons from the quaternary carbon atoms (C) shifted. A chemical movement was observed at δH = 2.31 and 1.62 ppm (2H, t, 7.8 Hz) for 2 protons of 2 CH2 groups used in forming triplet that interacted with 2 protons from the CH2 group. At δH = 1.29 and 1.31 ppm (2H, q, 7.8 Hz), 2 protons of 2 CH2 groups that formed quartet due to its reaction with 2 protons of the CH2 group and one proton of the CH group shifted, and at δH = 0.88 ppm (3H, t, 7.2 Hz), there was a chemical shift of 3 protons of CH3 group that interacted with the CH2 group.
Furthermore, the 1H-NMR spectrum of geranyl caprylate data revealed that at δH = 4.59 ppm (2H, d, 7.2 Hz), 2 protons (H) of the CH2 group that formed doublet as adjacent to CH group shifted and downfield because of its interaction with the electronegative oxygen and ester carbonyl (C=O) group. At δH = 5.40 and 5.18 ppm (1H, t, 7.2 Hz), one proton of 2 CH groups interacting with 2 protons from the CH2 group shifted and downfield because of its double bond. At dH = 2.08 and 2.09 ppm (2H, t, 7.2 Hz), 2 protons of 2 CH2 groups interacting with CH2 group shifted. At δH = 1.67 and 1.61 ppm (3H, s), 2 CH3 groups interacting with 2 protons from the quaternary carbon atoms (C) shifted. At δH = 2.29 and 1.67 ppm (2H, t, 7.8 Hz), 2 protons of 2 CH2 group used to form triplet and interacting with 2 protons from the CH2 group shifted. At δH = 1.29 and 1.28 ppm (2H, q, 7.8 Hz), 2 protons of 3 CH2 groups used to form quartet because of its interaction with 2 protons of the CH2 group and 1 proton of the CH group shifted, and at dH = 0.88 ppm (3H, t, 7.1 Hz), 3 protons of CH3 group interacting with CH2 group shifted.
Table 1: 1H-NMR data for geraniol esters (at 500 MHz in CDCl3, d in ppm, J in Hz).
|
Position (H) |
Geraniol |
Geranyl butyrate |
Geranyl caproate |
Geranyl caprylate |
|
3.65 (alcohol) |
|
|
|
|
|
1 |
4.18 (2, t) |
4.56 (2, d, 7.1) |
4.13 (2, d, 7.2) |
4.59 (2, d, 7.2) |
|
2 |
5.39 (1, t) |
5.41 (1, t, 7.2) |
5.39 (1, t, 7.3) |
5.40 (1, t, 7.2), |
|
3 |
– |
– |
– |
– |
|
4 |
2(2, t) |
2.16 (2, t, 7.2) |
2.06 (2, t, 7,1) |
2.09 (3, t, 7.2) |
|
5 |
2(2, t) |
2.16 (2, q, 7.2) |
2.01 (2, t, 7,2) |
2.08 (3, t, .2) |
|
6 |
5.2(1, t) |
5.31 (1, t, 7.2) |
5.09 (1, t, 7,3 ) |
5.18 (1, t, 7.2) |
|
7 |
– |
– |
– |
– |
|
8 |
1.82(3, s) |
1.67 (3, s,) |
1.67 (3, s) |
1.67 (3, s) |
|
9 |
1.70(3, s) |
1.51 (3, s) |
1.58 (3, s) |
1.61 (3, s) |
|
10 |
1.82(3, s) |
1.67 (3, s) |
1.67 (3, s) |
1.67 (3, s) |
|
1’ |
– |
– |
– |
– |
|
2’ |
– |
2.32 (2, t, 7.2) |
2.31 (2, t, 7,8) |
2.29 (2, t, 7.8) |
|
3’ |
– |
1.79 (2, m ,7.5) |
1.62 (2, t, 7,8) |
1.67 (2, t, 7.5) |
|
4’ |
– |
0.9 (3, t, 7.8) |
1.29 (2, q,7,8) |
1.29 (2, q, 7,8) |
|
5’ |
– |
– |
1.31 (2, q, 7.8) |
1.29 (2, q, 7.8) |
|
6’ |
– |
– |
0.88 (3, t, 7.2) |
1.29 (2, q, 7.8) |
|
7’ |
– |
– |
– |
1.28 (2, q, 7.5) |
|
8’ |
– |
– |
– |
0.88 (3, t, 7.1) |
Note: s : singlet, d : doublet, t : triplet, q : quartet, m : multiplet, and chemical shift of spectrum 1H-NMR of geraniol according to literature.
Cytotoxicity test is used in analyzing the degree of cell toxicity using human or animal cells. It is often used as a preliminary test in discovering new chemoprevention agents for cancer or anticancer drug candidates. For the purpose of this study, it was conducted through the use of BSLT method because it is easy, cheap, and reliable. The test showed that geraniol and geraniol esters were very active against A. salina with LC50 1.39 and 1.43-1.96 µg/ml as shown in Figure 3. They were active within LC50 value ≤ 30 µg/ml; therefore, they have the potentials of being chemo-preventive or anticancer agents.21,22
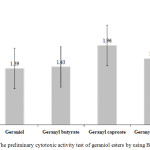 |
Figure 3: The preliminary cytotoxic activity test of geraniol esters by using BSLT method. |
The American National Cancer Institute (NCI) stated that murine leukemia (P388) cells are type of tumor cells that make use of cytotoxic test protocol. They are mostly used for further testing of compounds in order to check their suitability as chemo-preventive agents or anticancer drugs.27,28 Therefore, in vitro cytotoxic test of geraniol esters was conducted against murine leukemia (P388) cells using the MTT method. The result showed that geraniol esters have a cytotoxic effect and are active against murine leukemia (P388) cells (IC50<40 µg/mL28). The activity yielded IC50 22.34-32.29 µg/mL as shown in Figure 4. The IC50 shows the concentration of geraniol esters inhibiting 50% of the growth of murine leukemia (P388) cells, therefore, they have the prospects of being anticancer agents.
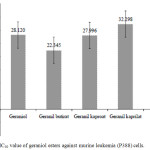 |
Figure 4: IC50 value of geraniol esters against murine leukemia (P388) cells. |
The IC50 values of geranyl butyrate and geranyl caproate were lower compared to geraniol. This means that they have higher anticancer activities, and through the esterification of geraniol, its activity can also improve. This is possible because of the increasing hydrophobicity or lipophilicity of geranyl butyrate and geranyl caproate compared to geraniol as indicated by increased log P of these compounds which are 3.75 and 4.62 for geranyl butyrate and geranyl caproate respectively and 2.49 for geraniol. Furthermore, increment in lipophilicity results in the variation of physical properties, for example, it increases its solubility in fat which consequently improves its power to penetrate cell membrane walls. It also causes an increment in the solubility of fluids that are not inside the cells which helps in improving how drugs are transported to the receptors. This research revealed that anticancer activity can be influenced by hydrophobicity or lipophilicity. This is in agreement with the result of a study conducted on the antitumoral properties of gallic acid and its ester derivatives which reported that tumor cell lines are more influenced by alkyl esters than garlic acid because of their hydrophobic mediety.29
The cytotoxic activity of geranyl caprylate (IC50 = 32 µg/ml) is lower than geraniol (IC50 = 28 µg/mL). This may be because of the high lipophilicity of geranyl caprylate (log P = 5.46 > 5). This makes it too soluble in fat, decrease its ability to penetrate the walls of the cell membrane and bioactivity. This is in accordance with the Lipinski’s rules five which states that a bioactive compound will be orally active if log P is less than 5 (Log P value of < 5).30-31
Artonin E, commonly used as a positive control of cytotoxic test against leukemia cancer cells, was used as the standard in testing the in vitro cytotoxic activity. The IC50 value of artonin E inhibiting 50% the growth of murine leukemia (P388) cancer cells was 0.4 µg/mL as shown in Figure 4. The IC50 of geraniol esters was high but lower than artonin E. This may be because of the low purity of the products, and the difference in their structures where geraniol esters are acyclic carbon compounds without amine content and artonin E is an aromatic carbon compound with amine content. Nevertheless, the result showed that acyclic compounds such as geraniol esters can be considered as new potential anticancer agents.
Furthermore, it was required to examine the cytotoxic activity of these geraniol esters against other cancer cells. Several previous studies reported that ester compounds can be used in the treatment of cancer. In the silico study of geraniol ester compounds like geranyl isobutyrate, geranyl benzoate, geranyl 3-methyl benzoate, geranyl 4-nitrobenzoate, and geranyl nicotinate, it was reported that their anti-inflammation potential is one of the causes of chronic diseases such as cancer in humans.32 Citronellyl 5-aminolevulinate, a citronellol ester compound derived from citronellol, has been reported to be effective in inhibiting cells growth of skin tumor. Cytotoxic activity of the compound on mice was found by photodynamic therapy to have increased significantly in deadly skin tumor cells about 10 times smaller than citronellol.33
Natural monoterpene ester compounds of essential oil such as linalyl acetate, geranyl acetate, and eugenol acetate were also reported to have the potential to be an antifungal, anesthetic, and anti-inflammatory which is all important in treating cancer.34 Geranyl acetate from the biotransformation of geraniol through esterase can be used as analgesic and anticancer.35,36 The geranium oil derived from Pelargonium geranium was also found to be active against certain cancers because of its ability to cause tumor regression. Citronellol, geraniol, and natural ester compounds such as citronellyl propanoate, geranyl propanoate, geranyl n-butyrate, and geranyl tiglate have all been observed to engage in anticancer activities.37 Paclitaxel (taxol) is also an anticancer compound derived from various Taxus species while one of the most commonly used chemotherapeutic agents against many types of cancer is tricyclic diterpene with a complex ester on the side chain.38-40 Therefore, geraniol esters which are acyclic compounds have the potentials of being the new alternative chemoprevention agents for cancer.
In vitro cytotoxicity against Vero cells was conducted through the use of the MTT method. The concentration of geraniol esters used for cytotoxicity test against Vero cells was 31.25-1000 µg/ml and three replicates were made in order to determine the 50% growth inhibition (IC50). This study showed that geraniol esters do not have any significant cytotoxicity on the normal Vero cell line as shown in Table 2.
Table 2: Inhibition percentage and IC50 geraniol esters against vero cell line.
|
Concentration of sample (µg/ml) |
Geranyl butyrate |
Geranyl caproate |
Geranyl caprylate |
|||
|
Inhibition (%)±SD |
IC50 (µg/ml) |
Inhibition (%)±SD |
IC50 (µg/ml) |
Inhibition (%)±SD |
IC50 (µg/ml) |
|
|
31.25 |
13.32±1.87 |
162.51 |
8.82±2.47 |
116.08 |
4.57±5.67 |
172.93 |
|
62.5 |
26.85±3.34 |
17.22±1.12 |
6.64±4.23 |
|||
|
125 |
25.29±3.59 |
51.54±3.91 |
18.49±2.65 |
|||
|
250 |
58.48±3.06 |
98.67±0.66 |
71.10±5.58 |
|||
|
500 |
91.99±1.95 |
101.17±0.16 |
101.33±0.78 |
|||
|
1000 |
97.61±1.43 |
101.22±0.32 |
102.34±0.16 |
|||
The result revealed that the inhibition of 92-101% and 98-102% was found at 500 and 1000 µg/ml respectively. These values indicate the cytotoxicity level of the geraniol esters such that the lower the IC50 values, the higher the toxicity, hence the cytotoxicity level of the samples was divided into strong (<100μg/ml), moderate (101-200 μg/ml), and weak (>200μg/ml). Geraniol esters were found to be indicating a weak inhibition against Vero cell lines.26
Conclusion
In conclusion, it was discovered that synthesizing geraniol esters through the use of NaOH as base catalyst produced geranyl butyrate, geranyl caproate, and geranyl caprylate with a medium yield (54-76%). The LC50 and IC50 values found against A. salina L. and murine leukemia (P388) cancer cells as well as their weak cytotoxic activity against Vero cells indicated their potential as anticancer agents. It was also found that the esterification of geraniol with butyric acid can enhance the anticancer activity of the compounds. Hydrophobicity or lipophilicity was also observed to have an influence on the anticancer activity such that increasing lipophilicity allows increasing anticancer activity, however in a situation where it is too lipophilic (Log P > 5), it may decrease. The study revealed that because of the ability of acyclic monoterpene esters to inhibit cancer, geraniol esters have the potentials of being the new alternative chemoprevention agents for cancer.
Acknowledgements
The research was funded by (Insinas Riset Pratama Individu (IRPI) Programme, grant number: 13/E/KPT/2018, cooperation agreement: 001/P/RPL-LIPI/INSINAS-2/VII/2018) the Ministry of Research, Technology, and Higher Education (Kemenristek-DIKTI) of the Republic of Indonesia. The authors express their gratitude to Professor Soleh Kosela and Dr Emil Budianto from the Department of Chemistry, Universitas Indonesia for their contribution to this article, and to Mrs. Evi Wulandari and Mr. Salahudin of the Research Center for Chemistry, Indonesian Institute of Sciences for their assistance in IR and GCMS analysis.
Conflict of Interest
There is no conflict of interest.
References
- Kamil, N.; Kamil, S. Systematic Rev. in Pharm. 2015, 6 (1), 13-17.
- Bernard, S.; and Christopher, P. IARC Nonserial Publication, WHO Press. 2014, 630.
- Anonymous. Pusat data dan informasi Depkes RI. 2015, 1-7.
- Newman, D.J.; and Cragg, G.M. J. of Nat. Prod. 2016: 79: 629-661.
- Qurishi, Y.; Hamid, A.; Majeed, R.; Hussain, A.; Qazi, A.K.; et. al. Future Oncol. 2011, 7 (8), 1007-1021.
- Mandal, S.; Bandyopadhyay, S.; Ghosh M.K.; Mandal, C. Anticancer Agent Med. Chem. 2012, 12, 49- 75.
- Christodoulu, M.; Kontos, C.; Halabalaki, M.; Scorilas, A. Anticancer Agent in Med. Chem. 2013, 13, 1-25.
- Ganjewala D. J. Of Essential Oil Therapeutics. 2009, 3, 56-65.
- Anonymous. National Agency for Export Development (NAFED). 2008, 1, 2.
- Raut, J.S.; Karuppayil, S.M. Ind. Crops. Prod. 2014, 62, 250-264.
- Cavar, S.; and Maksimovic, M. Food Control. 2012, 23, 263-267.
- Madnooni, M.; Mansour, K.; Gholivand, M. African J. Biotechnol. 2012, 11 (2), 498-503.
- Biblap, B.; and Jhinuk, C. APJCP. 2013, 14, 3735-3742.
- Can, B.K.H.; Buchbauer, G. Handbook of Essensial oil. Printed in the United States of America on acid-free paper. 2010, pp. 56.
- Chen, W.; Viljoen, A. South African J of Botany. 2010, 643- 651.
- Sharma, P.; Mondhe, D.; Muthiah, S. Chem. Biol. Interact. 2009, 179, 2 (3), 160-168.
- Bayala, B.; Bassole, I.; Simpore, J. Am. J. Cancer Res. 2014, 4 (6), 591-607.
- Fauser J. Anticancer Agent. Med. Chem. 2014, 14, 49-65.
- Narayanan, A.; Baskaran, S.A.; Amalaradjon, M.; Venkitanarayanan. Int. J. Mol. Sci. 2015, 16, 5014-5027.
- Widiyarti, G.; Hanafi, M.; Kosela, S.; Budianto, E. IJAC. 2016, 12 (3), 209-220.
- Meyer, B.R.; Ferrigni, N.R.; Putnam, J.E.: Jacobsen, L.B.; Nichols, D.E.; Mc. Laughlin, J.L. Planta Medica. 1982: 31-34.
- Kamanja, I.T.; Mbaria, J.M.; Gathumbi, P.K.; Mbaabu, M.B.; Kabasa, J.D.: Kiama, S.G. The J. of Med. Res. 2018, 4 (5), 249-255.
- Mosmann T. J .Immunol. Meth. 1983, 65, 55-63.
- Colegate, S.M.; Molyneux, R.J. Bioactive natural products: detection, isolation and structural determination. 2nd. edition. CRC Press Taylor and Francis Group. 2008. pp. 338-385.
- Khushboo, J.; Dhara, B.; Maitreyi, Z. Int. J. Pharm. Sci. Rev. Res. 2016, 37 (2), 130-133
- Garbi, M.I.; Osman, E.E.; Kabbashi, A.S.; Saleh, M.S. et. al. J. of Sci. and Innovative Res. 2015, 4 (2), 89-93.
- Dykes, D.J.; and Waud, W.R.; Cancer Research. 1997, 25-40.
- Ito,C.; Itoigawa, M.; Takakura, T.; Enjo, F.; Tokuda, H.; and Furukawa, H. J. of Nat. Prod. 2003: 66, 200-205.
- Locatelli, C.; Filippin, F.B.M. Eur. J. Med. Chem. 2013, 60, 233-239.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Freeney, F.J. Advanc. Drug Delivery Rev. 1997; 23, 3-25.
- Siswandono. Public Lecture medicinal chemistry: rational design of medicine and molecular modeling, Labtiap. BPPT Jakarta. 2015, 1-62.
- Ravi, S.; Sureshkumar, C.; Dhivya, S.; Dhamodaran, S.; and Narasimhan, S. Int. J. Curr .Res. 2012, 4 (9), 40-44.
- El-Far, M.; Elshal, M.; Bondock, S.; and Refaat, M. Int. J. Med. Sci. 2009, 1 (17), 278-288.
- De Sousa, D.P.; Júnior, G.A.S.; Andrade, L.N.; and Batista, J.S. Rec. Nat. Prod. 2011, 5, 117-122.
- Dipak, P.; Sikha, N.; Amit, B.; and Ahindra, N.; Int. J. Med. Chem. Sci. 2008, 6 (1), 11-16.
- De Sousa, D.P. Molecules. 2011, 16, 2233-2252.
- Saraswathi, J.; Venkatesh, K.; Nirmala, B.; Majid, H.H.; Rani, A.R.; J. Med. Plant Res. 2011, 5 (13), 2587-2598.
- Wang, Y.; and Tang, K. J. Biotechnol. 2011, 10 (72), 16379-16386.
- Priyadarshini, K.; and Keerthi, A.U.; Med. Chem. 2012, 2: 1-7.
- Barbuti, A.M.; and Chen, Z.S. Cancers. 2015; 7: 2360-2371.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


