Synthesis and Anticancer Activity Evaluation of Azepinobisindoles; the Isomeric Iheyamine A Derivatives
Weerachai Phutdhawong1, Sopita Rattanopas2, Jitnapa Sirirak2, Thongchai Taechowisan3 and Waya S. Phutdhawong*2
1Department of Science, Faculty of Liberal Arts and Science, Kasetsart University, Kamphang Sean campus, Nakhon Pathom 73140, Thailand.
2Department of Chemistry, Faculty of Science, Silpakorn University, Nakhon Pathom 73000, Thailand.
3Department of Microbiology, Faculty of Science, Silpakorn University, Nakhon Pathom 73000, Thailand.
Corresponding Author E-mail: waya.sengpracha@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/350231
Article Received on : 08-11-2018
Article Accepted on : 10-03-2019
Article Published : 08 Apr 2019
Azepinobisindole derivatives, the isomeric Iheyamine skeleton, was prepared and its anticancer activity evaluation were investigated against two human cancer cell lines, Hepatocellular carcinoma (HepG2) and human cervical cancer line (Hela) as well as the normal cell line (Vero cell line) using MTT assay. The anticancer activity results indicated that 2-methoxy-5-methyl-5H-azepino[2,3-b:4,5-bʹ]diindole was the most active derivative against tested cell lines. Additionally, molecular docking study in silico the possible inhibitory effect of cyclin-dependent kinase 2 (CDK2) by the azepinoindole revealed that all synthesized compounds fit well in the binding cavity of CDK2.
KEYWORDS:Anticancer Activity; Azepinobisindole; CDK2; Isomeric Iheyamine; Molecular Docking
Download this article as:| Copy the following to cite this article: Phutdhawong W, Rattanopas S, Sirirak J, Taechowisan T, Phutdhawong W. S. Synthesis and Anticancer Activity Evaluation of Azepinobisindoles; the Isomeric Iheyamine A Derivatives. Orient J Chem 2019;35(2). |
| Copy the following to cite this URL: Phutdhawong W, Rattanopas S, Sirirak J, Taechowisan T, Phutdhawong W. S. Synthesis and Anticancer Activity Evaluation of Azepinobisindoles; the Isomeric Iheyamine A Derivatives. Orient J Chem 2019;35(2). Available from: https://bit.ly/2IiFyIx |
Introduction
Bisindole alkaloids constitute an important class of secondary metabolites with characteristic cytotoxicity and pharmacological activities.1-5 Azepinobisindole alkaloids are a class of bisindole alkaloids containing a structural moiety of azepine (seven-membered ring) core, which demonstrated diverse biological activities especially anticancer effect. In the past years, number types of indole-fused azepines have been found with potential success in drug discovery. For example, Hyrtimomine A was reported to show anticancer activity against human epidermoid carcinoma KB cells and murine leukemia L1210 cells and showed antimicrobial activity against Candida albicans and Cryptococcus neoformans by Tanaka et al.6 Another example was Trigonoliimines C, which was reported with a modest anti-HIV-1 activity7 and their anticancer activity against human cervical cancer HeLa cells and human histiocytic lymphoma U-937 cells was also reported by Han et al.8 As illustrated in Fig 1, Iheyamine A (1) the azepinobisindole isolated from a colonial marine ascidian Polycitorella sp. by Sasaki et al. was reported to exhibit moderately cytotoxicity against murine leukemia P388 cells, human lung carcinoma A549 cells and human colon adenocarcinoma HT29 cells with IC50 in the level of 1 µg/mL.9 The synthesis of Iheyamine A was firstly accomplished by Lindsay et al. using cross-Mannich reaction.10 Recently, Abe and Yamada accomplished the synthesis of Iheyamine A via dehydrative Mannich-type reaction.11 Moreover, Bremner et al. synthesized the 5H-azepino[2,3-b:4,5-bʹ]-diindole system, which was the isomer of Iheyamine A.12 However, these structure motifs have not been studied for biological activities and their core structure resemble to Hytimomine A, which could be giving promising result for anticancer activity. Therefore, a series of 5H-azepino[2,3-b:4,5-bʹ]-diindole was synthesised and evaluated for cytotoxic activity. Molecular docking study against cyclic-dependent kinase 2 (CDK2) were also investigated the inhibitory ability of the designed compounds as well as the understanding of the antitumor results.
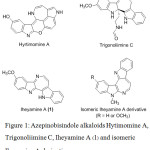 |
Figure 1: Azepinobisindole alkaloids Hytimomine A, Trigonoliimine C, Iheyamine A (1) and isomeric Iheyamine A derivatives. |
Materials and Methods
General Methods
Melting points were determined with a Stuart Scientific SMP 2 melting point apparatus and were uncorrected. FT-IR spectra were recorded using a Perkin Elmer Spectrum 100. NMR spectra were recorded on a Bruker Advance 300 (300 MHz for proton NMR and 75 MHz for carbon NMR) using tetramethylsilane (TMS) as an internal reference. Mass spectra were determined on a POLARIS Q or HEWLETT PACKARD 5973.
Synthesis Method
Isatin was purchased from FlukaTM and 5-Substituted isatins (2b-d) were prepared according to Sandmeyer isatin synthesis method.13
General Procedure for Synthesis of 3a–g
Isatin or 5-substituted isatin (7 mmol) and K2CO3 (10 mmol) were added to a dry CH3CN solution (25 mL) and stirred for 30 min at RT. Methyl iodide or benzyl chloride (7.5 mmol) was added to the solution and then refluxed at 80°C for 3 h. The mixture was cooled to RT and the solvent was removed. The residue was dissolved in CH2Cl2 (30 mL) and then washed with H2O (3×30 mL). The organic extracts were dried over anh Na2SO4 and concentrated to give the crude brown solid. The purification of the crude solid using flash-column chromatography (silica gel, hexane/EtOAc (2:1) gave the pure compounds 3a-g.
N–methylisatin (3a)
Orange solid; 62%; m.p. 128-129°C (lit.14129-130°C); IR (cm–1): 1721, 1603, 1466, 1365, 1325; 1H NMR (CDCl3): δ 3.26 (s, 3H, CH3), 6.90 (d, J = 8.2 Hz, 1H, ArH), 7.14 (t, J = 7.4 Hz, 1H, ArH), 7.58-7.64 (m, 2H, ArH); 13C NMR (CDCl3): δ 26.2, 109.9, 117.5, 123.9, 125.3, 138.4, 151.5, 158.3, 183.4.
5–methoxy–N–methylisatin (3b)
Red solid; 82%; m.p. 171-173°C (lit.15 175-176°C); IR (cm–1): 1722, 1625, 1485, 1356, 1287, 1226; 1H NMR (CDCl3): δ 3.23 (s, 3H, CH3), 3.81 (s, 3H, OCH3), 6.81 (d, J = 9.3 Hz, 1H, ArH), 7.14-7.18 (m, 2H, ArH) ); 13C NMR (CDCl3): δ 26.2, 56.0, 109.6, 110.9, 117.8, 124.6, 145.4, 156.6, 158.3, 183.8.
5–bromo–N–methylisatin (3c)
Orange solid; 70%; m.p. 167-169°C (lit.15 163-164°C); IR (cm–1): 1722, 1603, 1439, 1349, 1321, 711; 1H NMR (CDCl3): δ 3.24 (s, 3H, CH3), 6.80 (dd, J = 1.5, 7.4 Hz, 1H, ArH), 7.70 (s, 1H, ArH), 7.71 (dd, J = 1.8, 7.5 Hz, 1H, ArH); 13C NMR (CDCl3): δ 26.4, 111.6, 116.7, 118.6, 128.1, 140.6, 150.2, 157.5, 182.2.
5–nitro–N–methylisatin (3d)
Yellow solid; 76%; m.p. 195-197°C (lit.16 197-199 °C); IR (cm–1): 1742, 1607, 1515, 1467, 1326, 1291; 1H NMR (DMSO-d6): δ 7.37 (d, J = 8.8 Hz, 1H, ArH), 8.24 (d, J = 2.4 Hz, 1H, ArH), 8.55 (dd, J = 2.4, 8.7 Hz, 1H, ArH); 13C NMR (DMSO-d6): δ 27.0, 111.4, 118.3, 119.4, 133.5, 143.4, 156.1, 159.4, 181.7.
N–benzylisatin (3e)
Orange solid; 79%; m.p. 127-130°C (lit.17 131°C); IR (cm–1): 1728, 1608, 1468, 1347, 1307; 1H NMR (CDCl3): δ 4.94 (s, 2H, CH2), 6.78 (d, J = 8.0 Hz, 1H, ArH), 7.09 (td, J = 0.8, 7.6 Hz, 1H, ArH), 7.30-7.37 (m, 5H, ArH), 7.48 (td, J = 1.4, 7.8 Hz, 1H, ArH), 7.62 (dd, J = 0.8, 7.5 Hz, 1H, ArH); 13C NMR (CDCl3): δ 44.0, 111.0, 117.6, 123.9, 125.3, 127.4 (2C), 128.1 129.0 (2C), 134.5, 138.4, 150.7, 158.3, 183.3.
5–methoxy–N–benzylisatin (3f)
Red Solid; 52%; m.p. 123-124°C (lit.18 127-128°C); IR (cm–1): 1721, 1618, 1490, 1335, 1269, 1237; 1H NMR (CDCl3): δ 3.77 (s, 3H, OCH3), 4.91 (s, 2H, CH2), 6.67 (d, J = 8.6 Hz, 1H, ArH), 7.03 (dd, J = 2.7, 8.6 Hz, 1H, ArH), 7.15 (d, J = 2.7 Hz, 1H, ArH), 7.29-7.38 (m, 5H, ArH); 13C NMR (CDCl3): δ 44.0, 55.9, 109.6, 112.0, 118.1, 124.6, 127.4 (2C), 128.1, 129.0 (2C), 134.6, 144.6, 156.5, 158.4, 183.6.
5–bromo–N–benzylisatin (3g)
Orange solid; 92%; m.p. 101-103°C (lit.19 106-108°C); IR (cm-1): 1729, 1600, 1468, 1351, 1324, 696; 1H NMR (CDCl3): δ 4.93 (s, 2H, CH2), 6.67 (d, J = 8.4 Hz, 1H, ArH), 7.29-7.39 (m, 5H, ArH), 7.58 (dd, J = 2.1, 8.4 Hz, 1H, ArH), 7.72 (d, J = 2.1 Hz, 1H, ArH); 13C NMR (CDCl3): δ 44.2, 112.7, 116.8, 118.8, 127.4 (2C), 128.2, 128.4, 129.2 (2C), 134.0, 140.5, 149.4, 157.5, 182.1.
General Procedure for Synthesis of 5a–g
N-substituted isatin (3a–g) (0.9 mmol) and tryptamine (4) (1 mmol) in EtOH (5mL) in the presence of (0.2 mL) glacial acetic acid and 4 Å molecular sieve was stirred at RT for 24 h. The precipitate was removed by filtration and the filtrate was concentrated and purified by column chromatography (silica gel, hexane/EtOAc (2:1)) to give compound 5a–g.
Spiro[(1–methyl–3H–indole–2(1H)-one)-3,1ʹ-(1ʹ,2ʹ,3ʹ,4ʹ–tetrahydro–9H–pyrido[3,4–b]indol)] (5a)
Cream solid; 73%; m.p. 232-233°C (lit.12 239-241°C); IR (cm–1): 3139, 1714, 1609, 1469, 1347, 1296; 1H NMR (CDCl3): δ 2.95 (t, J = 5.7 Hz, 2H, CH2), 3.24 (s, 3H, CH3), 3.31 (dt, J = 5.4, 13.3 Hz, 1H, CH2), 3.81 (dt, J = 6.1, 13.3, 1H, CH2), 6.91 (d, J = 7.8, 1H, ArH), 7.03 (td, J = 0.9, 7.5, 1H, ArH), 7.08-7.12 (m, 3H, ArH), 7.17 (dd, J = 0.8, 7.8 Hz, 1H, ArH), 7.36 (td, J = 1.3, 7.7 Hz, 1H, ArH), 7.46 (br s, 1H, NH), 7.51-7.58 (m, 1H, ArH); 13C NMR (CDCl3): δ 21.1, 25.6, 39.0, 60.5, 107.7, 110.0, 111.5, 117.5, 118.6, 121.4, 122.3, 123.7, 126.2, 128.9, 129.1, 130.5, 135.2, 142.6, 175.6.
Spiro[(5–methoxy–1–methyl–3H–indole–2(1H)-one)-3,1ʹ-(1ʹ,2ʹ,3ʹ,4ʹ–tetrahydro–9H–pyrido[3,4–b]indol)] (5b)
Beige solid; 87%; m.p. 243-245°C (lit.12 255-256°C); IR (cm-1): 3292, 1708, 1600, 1496, 1341, 1296, 1231; 1H NMR (CDCl3): δ 2.98 (t, J = 5.7 Hz, 2H, CH2), 3.25 (s, 3H, CH3), 3.34 (dt, J = 5.4, 13.2 Hz, 1H, CH2), 3.72 (s, 3H, OCH3), 3.88 (dt, J = 6.3, 13.2 Hz, 1H, CH2), 6.81-6.91 (m, 3H, ArH), 7.08-7.19 (m, 3H, ArH), 7.29 (br s, 1H, NH), 7.57 (d, J = 5.9 Hz, 1H, ArH); 13C NMR (CDCl3): δ 22.0, 26.6, 40.0, 55.9, 62.0, 109.4, 111.2, 111.6, 111.9, 114.7, 118.4, 119.4, 122.3, 127.0, 129.9, 132.7, 136.4, 136.9, 156.6, 176.7.
Spiro[(5–nitro–1–methyl–3H–indole–2(1H)-one)-3,1ʹ-(1ʹ,2ʹ,3ʹ,4ʹ–tetrahydro–9H–pyrido[3,4–b]indol)] (5d)
Cream solid; 89%; m.p. 279-281°C; IR (cm–1): 3259, 1702, 1606, 1509, 1489, 1324, 1293; 1H NMR (CDCl3): δ 3.01 (t, J = 5.6, 2H, CH2), 3.35 (s, 3H, CH3), 3.40-3.43 (m, 1H, CH2), 3.79 (dt, J = 5.7, 13.5, 1H, CH2), 7.05 (d, J = 8.7, 1H, ArH), 7.09-7.19 (m, 3H, ArH), 7.59 (d, J = 6.4, 1H, ArH), 8.08 (d, J = 2.3, 1H, ArH), 8.35 (dd, J = 2.3, 8.7, 1H, ArH); 13C NMR (CDCl3): δ 21.9, 27.1, 61.1, 109.7, 111.6 (2C), 118.6, 119.1, 120.0, 122.1, 126.9, 127.0, 130.5, 133.5, 136.5, 143.1, 150.8, 177.6; HREI-MS (m/z) calculated for C19H17N4O3 (M+H)+ 349.1295, found 349.1308.
Spiro[(1–benzyl–3H–indole–2(1H)-one)-3,1ʹ-(1ʹ,2ʹ,3ʹ,4ʹ–tetrahydro–9H–pyrido[3,4–b]indol)] (5e)
Cream Solid; Yield 85%; m.p. 142-146°C; IR (cm–1): 3295, 1714, 1606, 1461, 1347, 1293; 1H NMR (CDCl3): δ 2.98 (t, J = 5.7 Hz, 2H, CH2), 3.37 (dt, J = 5.3, 13.2 Hz, 1H, CH2), 3.92 (dt, J = 6.2, 13.2 Hz, 1H, CH2), 4.86 (d, J = 15.5 Hz, 1H, CH2), 5.00 (d, J = 15.5 Hz, 1H, CH2), 6.85 (d, J = 7.8 Hz, 1H, ArH), 7.00 (td, J = 0.9, 7.5 Hz, 1H, ArH), 7.07-7.16 (m, 3H, ArH), 7.19 (d, J = 7.4 Hz, 1H, ArH), 7.25 (td, J = 1.3, 7.8 Hz, 1H, ArH), 7.29-7.40 (m, 5H, ArH), 7.54-7.61 (m, 1H, ArH); 13C NMR (CDCl3): δ 22.1, 40.1, 44.0, 61.5, 109.6, 111.0, 112.6, 118.6, 119.7, 122.5, 123.4, 124.8, 127.3, 127.6 (2C), 127.9, 129.0 (2C), 129.7, 130.2, 131.5, 135.8, 136.3, 142.6, 176.9; HREI-MS (m/z) calculated for C25H22N3O (M+H)+ 380.1757, found 380.1761.
Spiro[(5–bromo–1–methyl–3H–indole–2(1H)-one)-3, 1ʹ-(1ʹ,2ʹ,3ʹ,4ʹ–tetrahydro–9H–pyrido[3,4–b]indol)] (5c)
Beige solid; Yield 89%; m.p. 240-242°C; IR (cm–1): 3267, 1708, 1607, 1459, 1348, 1229, 688; 1H NMR (CDCl3): δ 2.98 (t, J = 5.7 Hz, 2H, CH2), 3.25 (s, 3H, CH3), 3.32 (dt, J = 5.5, 13.2 Hz, 1H, CH2), 3.83 (dt, J = 5.9, 13.2 Hz, 1H, CH2), 6.82 (d, J = 8.3 Hz, 1H, ArH), 7.09-7.20 (m, 3H, ArH), 7.30 (br s, 1H, NH), 7.34 (d, J = 1.9 Hz, 1H, ArH), 7.50 (dd, J = 2.0, 8.3 Hz, 1H, ArH), 7.56 (d, J = 6.5 Hz, 1H, ArH); 13C NMR (CDCl3): δ 19.8, 24.4, 37.8, 59.3, 108.0, 108.8, 110.6, 113.7, 116.4, 117.5, 120.5, 124.8, 125.7, 126.9, 130.4, 131.2, 134.0, 140.3, 173.9; HREI-MS (m/z) calculated for C19H17BrN3O (M+H)+ 382.0550, found 382.0555.
Spiro[(5–methoxy–1–benzyl–3H–indole–2(1H)-one)-3,1ʹ-(1ʹ,2ʹ,3ʹ,4ʹ–tetrahydro–9H–pyrido[3,4–b]indol)] (5f)
Beige solid; Yield 84%; m.p. 180-183°C; IR (cm–1): 3290, 1601, 1710, 1494, 1336, 1229, 1202; 1H NMR (CDCl3): δ 2.99 (t, J = 5.7 Hz, 2H, CH2), 3.36 (dt, J = 5.3, 13.1 Hz, 1H, CH2), 3.66 (s, 3H, OCH3), 3.95 (dt, J = 6.3, 13.1 Hz, 1H, CH2), 4.83 (d, J = 15.4 Hz, 1H CH2), 5.0 (d, J = 15.4 Hz, 1H, CH2), 6.75-6.80 (m, 3H, ArH), 7.09-7.18 (m, 3H, ArH), 7.30 (br s, 1H, NH), 7.31-7.36 (m, 5H, ArH), 7.58 (d, J = 6.9 Hz, 1H ArH); 13C NMR (CDCl3) δ 22.0, 40.1, 44.0, 55.7, 61.9, 110.2, 111.1, 111.3, 112.5, 114.8, 118.6, 119.6, 122.5, 127.2 (2C), 127.5, 127.9, 128.9 (2C), 130.2, 132.6, 135.7, 135.9, 136.3, 156.5, 176.7; HREI-MS (m/z) calculated for C26H24N3O2 (M+H)+ 410.1863, found 410.1873.
Spiro[(5–bromo–1–benzyl–3H–indole–2(1H)-one)-3,1ʹ-(1ʹ,2ʹ,3ʹ,4ʹ–tetrahydro–9H–pyrido[3,4–b]indol)] (5g)
Cream solid; Yield 75%; m.p. 135-138°C; IR (ATR) 3323, 1719, 1603, 1477, 1351, 1229, 1170, 691; 1H NMR (CDCl3): δ 2.99 (t, J = 5.7, 2H Hz, CH2), 3.35 (dt, J = 5.4, 13.2 Hz, 1H, CH2), 3.88 (dt, J = 6.1 Hz, 1H, CH2), 4.85 (d, J = 15.5 Hz, 1H, CH2), 4.99 (d, J = 15.5 Hz, 1H, CH2), 6.72 (d, J = 8.3 Hz, 1H, ArH), 7.08-7.20 (m, 3H, ArH), 7.22 (br s, 1H, NH), 7.30-7.45 (m, 7H, ArH), 7.58 (d, J = 6.4 Hz, 1H, ArH); 13C NMR (CDCl3): δ 21.7, 39.8, 43.7, 61.2, 110.8, 110.9, 112.5, 115.8, 118.4, 119.5, 122.4, 126.8, 127.1 (2C), 127.7 (2C), 128.7 (2C), 129.0, 132.2, 133.2, 134.9, 136.0, 141.2, 176.0; HREI-MS (m/z) calculated for C25H21BrN3O (M+H)+ 458.0863, found 458.0863.
General Procedure for Synthesis of 7a–g
A solution of spiroindolone 5a-g (0.3 mmol) in dry toluene (6 mL) was stirred and cooled to -78°C under Ar atmosphere and DIBAL-H (3 mmol) was added dropwise into the solution. After 4 h, a solution of 5% aqueous NaOH (8 mL) was quenched to the mixture and extracted with EtOAc (3×15 mL). The organic extracts were combined and washed with brine, dried over anhydrous sodium sulfate and after that concentrated under reduced pressure to give oxyindoline 6a-g. (This compound was unstable and used without further purification due to the color changed on a silica gel and in CDCl3.)
The oxyindoline 6a-g was warmed in 5M HCl (10 mL) for 1 h at 90°C and then basified with 25% KOH until the pH was more than 12. The solid was filtered and purified by column chromatography (silica gel) to give isomeric Iheyamine derivatives 7a-g.
5–methyl–5H–azepino[2,3–b:4,5–bʹ]diindole (7a)
Following the general procedure, spiro[(1-methyl-3H-indole-2(1H)-ol)-3,1ʹ-(1ʹ,2ʹ,3ʹ,4ʹ-tetrahydro-9H-pyrido[3,4-b]indol)] (6a) was obtained as light brown viscous oil; 1H NMR (CDCl3): δ 2.86 (t, J = 5.6 Hz, 2H, CH2), 2.93 (s, 3H, CH3), 3.07 (dt, J =6.3, 12.8 Hz, 1H, CH2), 3.42 (dt, J = 5.0, 12.9 Hz, 1H, CH2), 4.92 (s, 1H, CH), 6.60 (d, J = 7.8 Hz, 1H, ArH), 6.69 (td, J = 0.8, 7.4 Hz, 1H, ArH), 6.92 (dd, J = 0.9, 7.4 Hz, 1H, ArH), 7.07-7.16 (m, 2H, ArH), 7.17-7.23 (m, 2H, ArH), 7.52 (dd, J = 1.7, 6.6 Hz, 1H, ArH), 7.72 (br s, 1H, NH).
The rearrangement of oxyindole 6a in 5M HCl gave compound 7a as green solid; 13 % (2 steps); m.p. 194-197°C (lit.12 193-195°C); IR (cm-1): 2921, 2852, 1547, 1468, 1365; 1H NMR (CDCl3): δ 4.25 (s, 3H, CH3), 7.45 (t, J = 7.8 Hz, 1H, ArH), 7.57-7.72 (m, 2H, ArH), 7.75 (d, J = 8.1 Hz, 1H, ArH), 7.83 (t, J = 8.1 Hz, 1H, ArH), 8.13 (d, J = 8.3 Hz, 1H, ArH), 8.35 (d, J = 7.8 Hz, 1H, ArH), 8.48 (d, J = 5.8 Hz, 1H, CH), 9.03 (d, J = 5.8 Hz, 1H, CH), 9.46 (d, J = 7.9 Hz, 1H, ArH); 13C NMR (CDCl3): δ 28.8, 109.4, 112.3, 119.5, 121.6 (2C), 122.3, 122.8, 126.0, 126.5, 127.6, 128.7, 132.6, 139.9, 145.5, 147.2, 147.4, 156.2, 160.2; HREI-MS (m/z) calculated for C20H16N3O (M+H)+ 284.1182, found 284.1178.
2–Methoxy–5–methyl–5H–azepino[2,3–b:4,5–bʹ]diindole (7b)
Following the general procedure, Spiro[(5-methoxy-1-methyl-3H-indole-2(1H)-ol)-3,1ʹ-(1ʹ,2ʹ,3ʹ,4ʹ-tetrahydro-9H-pyrido[3,4-b]indol)] (6b) was obtained as brown viscous oil; 1H NMR (CDCl3): δ 2.86 (t, J = 4.9 Hz, 2H, CH2), 2.87 (s, 3H, CH3), 3.08 (dt, J = 6.1, 12.7 Hz, 1H, CH2), 3.39 (dt, J = 5.2, 12.8 Hz, 1H, CH2), 3.66 (s, 3H, OCH3), 4.86 (s, 1H, CH), 6.53 (d, J = 8.7 Hz, 1H, ArH), 6.56 (d, J = 2.5 Hz, 1H, ArH), 6.79 (dd, J = 2.6, 8.4 Hz, 1H, ArH), 7.09-7.23 (m, 3H, ArH), 7.52 (d, J = 7.5 Hz, 1H, ArH), 7.79 (br s, 1H, NH).
The rearrangement of oxyindole 6b in 5M HCl gave compound 7b as Burgundy solid; 14% (2 steps); m.p. 148-151°C (lit.12 147-148°C); IR (cm–1): 2922, 2852, 1539, 1497, 1369, 1261; 1H NMR (CDCl3): δ 4.15 (s, 3H, CH3), 4.23 (s, 3H, OCH3), 7.42 (dd, J = 2.6, 8.9 Hz, 1H, ArH), 7.43 (t, J = 7.5 Hz, 1H, ArH), 7.60 (d, J = 8.9 Hz, 1H, ArH), 7.81 (t, J = 7.7 Hz, 1H, ArH), 8.11 (d, J = 8.0 Hz, 1H, ArH), 8.32 (d, J = 7.7 Hz, 1H, ArH), 8.43 (d, J = 5.7 Hz, 1H, CH), 8.97 (d, J = 5.8 Hz, 1H, CH), 9.12 (d, J = 2.5 Hz, 1H, ArH); 13C NMR (CDCl3): δ 26.7, 54.1, 105.1, 108.1, 109.5, 116.9, 117.7, 118.2, 119.2, 119.5, 120.5, 123.4, 130.2, 132.9, 142.8, 143.9, 145.3, 153.0, 153.8, 156.4; HREI-MS (m/z) calculated for C19H14N3 (M+H)+ 314.1288, found 314.1286.
2–Bromo–5–methyl–5H–azepino[2,3–b:4,5–bʹ]diindole (7c)
Following the general procedure, spiro[(5-bromo-1-methyl-3H-indole-2(1H)-ol)-3,1ʹ-(1ʹ,2ʹ,3ʹ,4ʹ-tetrahydro-9H-pyrido[3,4-b]indol)] (6c) was obtained as brown viscous oil; 1H NMR (CDCl3): δ 2.86 (t, J = 5.6 Hz, 2H, CH2), 2.90 (s, 3H, CH3), 3.06 (dt, J = 6.3, 12.8 Hz, 1H, CH2), 3.39 (dt, J = 5.0, 12.9 Hz, 1H, CH2), 4.91 (s, 1H, CH), 6.46 (d, J = 8.3 Hz, 1H, ArH), 7.01 (d, J = 2.0 Hz, 1H, ArH), 7.11 (td, J = 1.4, 7.1 Hz, 1H, ArH), 7.16 (td, J = 1.5, 7.3 Hz, 1H, ArH), 7.23 (d, J = 7.0 Hz, 1H, ArH), 7.31 (dd, J = 2.0, 8.3 Hz, 1H, ArH), 7.53 (d, J = 7.2 Hz, 1H, ArH), 7.70 (br s, 1H, ArH).
The rearrangement of oxyindole 6c in 5M HCl gave compound 7c as green solid; 14% (2 steps); m.p. 213-215°C; IR (cm–1): 2921, 2852, 1551, 1463, 1369, 589; 1H NMR (CDCl3): δ 4.18 (s, 3H, CH3), 7.45 (t, J = 7.9 Hz, 1H, ArH), 7.49 (d, J = 8.8 Hz, 1H, ArH), 7.79 (dd, J = 2.0, 8.7 Hz, 1H, ArH), 7.83 (t, J = 7.7 Hz, 1H, ArH), 8.10 (d, J = 8.1 Hz, 1H, ArH), 8.30 (d, J = 7.8 Hz, 1H, ArH), 8.37 (d, J = 5.7 Hz, 1H, CH), 8.98 (d, J = 5.7 Hz, 1H, CH), 9.70 (d, J = 2.0 Hz, 1H, ArH); 13C NMR (CDCl3): δ 27.0, 108.8, 113.3, 117.7, 117.8, 118.5, 119.8, 120.0, 120.9, 121.9, 123.9, 125.6, 127.4, 129.3, 130.9, 136.2, 137.8, 144.4, 145.3; HREI-MS (m/z) calculated for C19H13BrN3 (M+H)+ 362.0287, found 362.0283.
2–nitro–5–methyl–5H–azepino[2,3–b:4,5–bʹ]diindole (7d)
Following the general procedure, spiro[(5-nitro-1-methyl-3H-indole-2(1H)-ol)-3,1ʹ-(1ʹ,2ʹ,3ʹ,4ʹ-tetrahydro-9H-pyrido[3,4-b]indol)] (6d) was obtained as brown viscous oil; 1H NMR (CDCl3): δ 2.85-2.91 (m, 2H, CH2), 3.01-3.12 (m with s prominent, 4H, CH2, CH3), 3.45 (dt, J = 4.6, 13.0 Hz, 1H, CH2), 5.11 (s, 1H, CH), 6.49 (d, J = 8.9 Hz, 1H, ArH), 7.11-7.23 (m, 3H, ArH), 7.55 (d, J = 6.9 Hz, 1H, ArH), 7.60 (br s, 1H, NH), 7.78 (d, J = 2.3 Hz, 1H, ArH), 8.17 (dd, J = 2.3, 8.8 Hz, 1H, ArH).
The rearrangement of oxyindole 6d in 5M HCl gave compound 7d as green solid; 10% (2 steps); m.p. 328-330°C; IR (cm–1): 2922, 2852, 1554, 1506, 1455, 1307, 1293; 1H NMR (CDCl3): δ 4.24 (s, 3H, CH3), 7.49 (t, J = 7.3 Hz, 1H, ArH), 7.65 (d, J = 8.8 Hz, 1H, ArH), 7.87 (t, J = 8.2 Hz, H, ArH), 8.15 (d, J = 8.0 Hz, 1H, ArH), 8.32 (d, J = 7.4 Hz, 1H, ArH), 8.42 (d, J = 5.7 Hz, 1H, CH), 8.57 (dd, J = 2.4, 9.2 Hz, 1H, ArH), 9.04 (d, J = 5.6 Hz, 1H, CH), 10.43 (d, J = 2.1 Hz, 1H, ArH); 13C NMR (CDCl3): δ 29.8, 110.7, 112.0, 117.9, 118.5, 118.6, 121.1, 122.1, 123.7, 124.4, 125.4, 126.0, 126.6, 127.7, 130.7, 136.9, 141.3, 143.8, 151.3.; HREI-MS (m/z) calculated for C19H12N4O2 (M+H)+ 329.1039, found 329.1041.
5–benzyl–5H–azepino[2,3–b:4,5–bʹ]diindole (7e)
Following the general procedure, spiro[(1-benzyl-3H-indole-2(1H)-ol)-3,1ʹ-(1ʹ,2ʹ,3ʹ,4ʹ-tetrahydro-9H-pyrido[3,4-b]indol)] (6e) was obtained as light brown viscous oil; 1H NMR (CDCl3): δ 2.81-2.88 (m, 2H, CH2), 3.00-3.09 (m, 1H, CH2), 3.38-3.46 (m, 1H, CH2), 4.41 (d, J = 15.2 Hz, 1H, CH2), 4.58 (d, J = 15.1 Hz, 1H, CH2), 5.05 (s, 1H, CH), 6.58 (d, J = 7.9 Hz, 1H, ArH), 6.67 (td, J = 0.8, 7.5 Hz, 1H, ArH), 6.93 (dd, J = 0.9, 7.4 Hz, 1H, ArH), 7.05-7.21 (m, 4H, ArH), 7.27-7.39 (m, 3H, ArH), 7.44 (d, J = 6.7 Hz, 2H, ArH), 7.51 (d, J = 7.5 Hz, 1H, ArH), 7.64 (br s, 1H, NH).
The rearrangement of oxyindole 6e in 5M HCl gave compound 7e as Green solid; Yield 11% (2 steps); m.p. 143-146°C; IR (cm-1): 2923, 2851, 1609, 1454, 1351; 1H NMR (CDCl3): δ 6.04 (s, 2H, CH2), 7.17-7.35 (m, 5H, ArH), 7.46 (t, J = 7.5 Hz, 1H, ArH), 7.55-7.72 (m, 3H, ArH), 7.83 (t, J = 7.6 Hz, 1H, ArH), 8.14 (d, J = 8.1 Hz, 1H, ArH), 8.35 (d, J = 7.7 Hz, 1H, ArH), 8.66 (d, J = 5.7 Hz, 1H, CH), 9.02 (d, J = 5.7 Hz, 1H, CH), 9.65 (d, J = 7.7 Hz, 1H, ArH); 13C NMR (CDCl3): δ 43.3, 107.7, 110.0, 117.0, 117.5, 119.3, 119.4, 120.1, 120.5, 123.6, 124.4 (2C), 124.9, 125.1, 126.2 (2C), 126.4, 130.3, 134.7, 137.0, 143.5, 144.9, 145.0, 153.9, 157.6; HREI-MS (m/z) calculated for C25H18N3 (M+H)+ 360.1495, found 360.1485.
2–methoxy–5–benzyl–5H–azepino[2,3–b:4,5–bʹ]diindole (7f)
Following the general procedure, spiro[(5-methoxy-1-benzyl-3H-indole-2(1H)-ol)-3,1ʹ-(1ʹ,2ʹ,3ʹ,4ʹ-tetrahydro-9H-pyrido[3,4-b]indol)] (6f) was obtained as brown viscous oil; 1H NMR (CDCl3): δ 2.81-2.90 (m, 2H, CH2), 2.98-3.11 (m, 1H, CH2), 3.35-3.45 (m, 1H, CH2), 3.64 (s, 3H, OCH3), 4.33 (d, J = 14.9 Hz, 1H, CH2), 4.52 (d, J = 14.8 Hz, 1H, CH2), 5.01 (s, 1H, CH), 6.48 (d, J = 8.5 Hz, 1H, ArH), 6.56 (d, J = 2.6 Hz, 1H, ArH), 6.71 (dd, J = 2.6, 8.5 Hz, 1H, ArH), 7.06-7.22 (m, 3H, ArH), 7.27-7.45 (m, 5H, ArH), 7.51 (d, J = 7.5 Hz, 1H, ArH), 7.71 (br s, 1H, NH).
The rearrangement of oxyindole 6f in 5M HCl gave compound 7f as burgundy solid; 13% (2 steps); m.p. 209-211°C; IR (cm–1): 2923, 2852, 1544, 1497, 1314, 1268; 1H NMR (CDCl3): δ 4.11 (s, 3H, OCH3), 5.97 (s, 2H, CH2), 7.18-7.24 (m, 5H, ArH), 7.29 (dd, J = 2.6, 8.9 Hz, 1H, ArH), 7.38-7.49 (m, 2H, ArH), 7.80 (t, J = 7.6 Hz, 1H, ArH), 8.11 (d, J = 8.0 Hz, 1H, ArH), 8.29 (d, J = 7.7 Hz, 1H, ArH), 8.38 (d, J = 5.7 Hz, 1H, ArH), 8.91 (d, J = 5.7 Hz, 1H, ArH), 9.09 (d, J = 2.5 Hz, 1H, ArH); 13C NMR (CDCl3): δ 44.1, 54.4, 105.6, 109.3, 110.2, 117.4, 118.3, 118.6, 119.8, 120.1, 121.4, 124.1, 124.9 (2C), 125.7, 126.2, 126.7, 126.9 (2C), 130.8, 132.8, 135.3, 143.5, 145.0, 145.6, 154.2; HREI-MS (m/z) calculated for C26H20N3O (M+H)+ 390.1600, found 360.1595.
2–Bromo–5–benzyl–5H–azepino[2,3–b:4,5–bʹ]diindole (7g)
Following the general procedure, spiro[(5-bromo-1-benzyl-3H-indole-2(1H)-ol)-3,1ʹ-(1ʹ,2ʹ,3ʹ,4ʹ-tetrahydro-9H-pyrido[3,4-b]indol)] (6g) was obtained as red viscous oil; 1H NMR (CDCl3): δ 2.81-2.91 (m, 2H, CH2), 3.04 (dt, J = 6.4, 13.0 Hz, 1H, CH2), 3.40 (dt, J = 4.8, 12.9 Hz, 1H, CH2), 4.39 (d, J = 15.1 Hz, 1H, CH2), 4.56 (d, J = 15.1 Hz, 1H, CH2), 5.05 (s, 1H, CH), 6.44 (d, J = 8.4 Hz, 1H, ArH), 7.02 (d, J = 2.0 Hz, 1H, ArH), 7.08-7.23 (m, 4H, ArH), 7.28-7.45 (m, 5H, ArH), 7.52 (d, J = 7.9 Hz, 1H, ArH), 7.53 (br s, 1H, NH).
The rearrangement of oxyindole 6g in 5M HCl gave compound 7g as green solid; 19% (2 steps); m.p. 213-216°C; IR (cm–1): 2921, 2846, 1551, 1450, 1372, 688; 1H NMR (CDCl3): δ 5.92 (s, 2H, CH2), 7.14-7.25 (m, 5H, ArH), 7.35 (d, J = 8.7 Hz, 1H, ArH), 7.44 (t, J = 7.7 Hz, 1H, ArH), 7.65 (dd, J = 1.5, 8.7 Hz, 1H, ArH), 7.82 (t, J = 7.6 Hz, 1H, ArH), 8.10 (d, J = 8.0 Hz, 1H, ArH), 8.26 (d, J = 7.6 Hz, 1H, ArH), 8.32 (d, J = 5.7 Hz, 1H, CH), 8.93 (d, J = 5.7 Hz, 1H, CH), 9.69 (s, 1H, ArH); 13C NMR (CDCl3): δ 44.2, 109.9, 110.8, 113.9, 117.7, 118.5, 120.0, 120.5, 122.2, 123.9, 125.1 (2C), 126.0, 127.0 (3C), 127.5, 129.8, 131.3, 135.0, 135.9, 145.2, 145.5, 153.6, 157.5; HREI-MS (m/z) calculated for C25H17BrN3 (M+H)+ 438.0600, found 438.0593.
Anticancer Activity
Cell Culture
Herein, human cervical cancer cells (HeLa), human hepatocellular carcinoma cells (HepG2) and kidney epithelial cells from African green monkey (Vero) were cultured in 10% Fetal Bovine Serum (FBS supplemented DMEM (Dulbecco’s Modified Eagle’s Medium) and antibiotics. Cells were kept in a humidified 5% CO2 atmosphere at 37 °C with medium changes every 2 days until cells grown to 80-85% confluency. They were collected by trypsinization procedure with 0.25% trypsin and the viable cells were counted by tryphan blue exclusion.
Determination of Anticancer Activity by MTT Assay
Cells were seeded on 96-well plate at a density of 1×105 cells/mL. After 24 h, the cells were administered with the test compounds at various concentrations and 20% organic solvent (acetone or DMSO) was used as a positive control. After incubation for 24 h, 100 µL of MTT (400 µg/mL) was added to each well and further incubated for 4 h. The medium was then removed and 100 µL of DMSO, which was used to dissolve the resulting formazan crystal, was added to each well. The microplate spectrometer (biochrom EZ Read 2000) was used to measure the absorbance at 540 nm. Percentage cell viability was calculated using corrected absorbance with the following equation:
% viable cells = [(Abssample – Absblank)/(Abscontrol – Absblank)] x 100
where Abssample was the absorbance of the test compound, Abscontrol was the absorbance of 1% organic solvent in medium and Absblank was the absorbance of 20% organic solvent in medium. The IC50 value was determined by plotting percentage cell viability versus sample concentration.
Results and Discussion
Chemistry
The isatin (2a) was purchased from Sigma-Aldrich and the 5-substituted isatins (2b–d) were prepared by classical Sandmeyer isatin synthesis methods.13 N-alkylation of isatins (2a–d) with alkyl halide in the presence of K2CO3 were completed the N-substituted isatin derivatives (5a–g) in 52-92% yield (scheme 1).
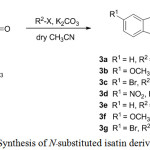 |
Scheme 1: Synthesis of N-substituted isatin derivatives (3a–g). |
The condensation reaction of N-substituted isatin derivatives (3a-g) with tryptamine (4) under acidic conditions forming the iminium ion intermediate, followed by spiro cyclization to provide racemic spiroindolone (5a-g) in good yield. Based on literature, the partial reduction of spiroindolones were achieved cleanly to the corresponding carbinolamines using sodium bis(2-methoxyethoxy)aluminium hydride.20 However, the partial reduction using DIBAL-H in handed gave the carbinolamine (6a-g) in low yields. The carbinolamine (6a-g) were used without further purification by heating in 5 M HCl at 90°C to undergo rearrangement to the azepinodiindole (7a-g) and yields were reported from a 2-step process. The structure of compounds (7a-g) were compared to the previous synthesis isomeric Iheyamines12 (scheme 2).
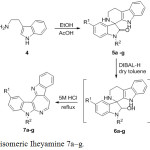 |
Scheme 2: Synthesis of isomeric Iheyamine 7a-g. |
Anticancer Activity
The in vitro anticancer activity of the synthesized compounds (7a-g) were evaluated for cytotoxicity against HeLa, HepG2, and Vero cell lines by MTT assay.21 All of the tested compounds exhibited good activity against both cancer cell lines and the IC50 values were shown in Table 1. The N-methyl derivative of azepinodiindoles (7a-c) were more potent than the N-benzyl derivatives (7e-g). The methoxy substituent on the aromatic ring of compound (7b) and bromine substituent (7c) had similar inhibitory effect against HepG2 and HeLa cell lines with the same IC50 value of 15.91 µg/mL. However, compound (7b) had lower activity on normal cell lines (Vero cell lines). The unsubstituted aromatic ring of azepinodiindoles (7a) and (7e) showed the selective inhibitory effect against HepG2 cell lines with no effect from the R2 substitution. On the aromatic ring, the electron withdrawing nitro group, (7d) was the least active compound compared with other compounds containing methyl groups on R2.
Table 1: Cytotoxicity of test compounds against cancer cell lines (HeLa and HepG2) and normal cell (Vero).
| Compounds | R1 | R2 |
IC50 (µg/mL) |
||
| HeLa | HepG2 | Vero | |||
| 7a | H | CH3 | 71.30 ± 1.11 | 27.27 ± 0.38 | 101.01 ± 0.15 |
| 7b | OCH3 | CH3 | 15.91 ± 0.36 | 22.62 ± 0.22 | 117.87 ± 0.37 |
| 7c | Br | CH3 | 15.91 ± 0.30 | 30.46 ± 0.83 | 43.30 ± 0.52 |
| 7d | NO2 | CH3 | 66.63 ± 0.61 | 95.07 ± 0.36 | 205.48 ± 0.26 |
| 7e | H | Bn | 61.01 ± 0.99 | 28.57 ± 0.48 | 75.99 ± 0.76 |
| 7f | OCH3 | Bn | 82.61 ± 0.47 | 63.00 ± 0.42 | 330.50 ± 0.72 |
| 7g | Br | Bn | 149.56 ± 0.29 | 122.97 ± 0.14 | 234.38 ± 0.29 |
| Acridine orange | – | – | 4.11 | 6.36 | 5.82 |
| Actinomycin D | – | – | NT | 2.70 | NT |
NT: not tested
Molecular Docking
Using the iGEMDOCK v2.1 software22, molecular docking was performed in order to explore binding positions along with intermolecular interactions between compounds (7a–g) and binding site of cyclin-dependent kinase 2 (CDK2) which was key protein for cell cycle progression.23,24 Compounds (7a–g) were docked into the active site of CDK2 co-crystallized with LZ8 (PDB ID: 2VTO). LZ8 was also redocked into the CDK2 and its binding energy and hydrogen bond was then compared to those of azepinodiindoles (7a–g). The docking results revealed that the binding position of redocked LZ8 (shown in orange in Fig 2) was more or less the same to that of co-crystallized LZ8 in CPX2. Table 2 showed that the binding energy of all synthesized compounds (7a–g) were -103.98 to -84.35 kcal/mol, which were higher than that of LZ8. Among these, compound 7b fit well in the binding cavity of CPK2 and had the most similar orientation and binding position to those of LZ8, as illustrated in Fig. 2. Additionally, this compound had three hydrogen bonding interaction with LEU83, LYS89 and GLN131. For 7a, 7c and 7d, it was found that they fit in the binding site of CPK2, however their orientation and binding pose were rather different to those of LZ8 (Fig. 3a). In contrast, although the azepinodiindoles (7e–g) containing Bn group for R2 substituent had lower binding energy than 7b, they could not fit in the binding site of CDK2, especially 7g.
Table 2: Summary of binding energy, amino acid interactions and hydrogen bond length of azepenodiindoles (7a–g) in binding site of CDK2.
| Compounds | Binding energy (kcal/mol) | Amino acid residue | Hydrogen bond length (Å) |
| 7a | -84.35 | ILE10, GLY11 | 2.48, 2.47 |
| 7b | -89.74 | LEU83, LYS89, GLN131 | 2.28, 2.39, 2.42 |
| 7c | -86.07 | – | – |
| 7d | -103.98 | ILE10, GLU12, LYS129 | 2.45, 2.30, 1.63 |
| 7e | -100.44 | ILE10, GLY11 | 2.84, 2.73 |
| 7f | -99.11 | GLU12, THR14, GLN131 | 2.98, 2.62, 2.89 |
| 7g | -101.47 | ILE10 | 2.69 |
| LZ8 | -114.60 | PHE82, LEU83 | 2.61, 2.21 |
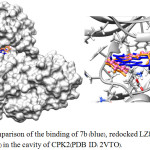 |
Figure 2: Comparison of the binding of 7b (blue), redocked LZ8 (orange) and LZ8 (pink) in the cavity of CPK2(PDB ID: 2VTO). |
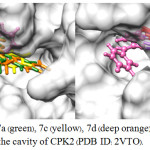 |
Figure 3: The binding of 7a (green), 7c (yellow), 7d (deep orange), 7e (red), 7f (light blue), 7g (purple) and LZ8 (pink) in the cavity of CPK2 (PDB ID: 2VTO).Click here to view figure |
Conclusion
In summary, syntheses of isomeric Iheyamine A derivatives 9a–g can be prepared from the synthetic isatin derivatives and commercially available tryptamine. There have been achieved in 3-11% overall yield with 4-6 steps. The key reactions including Sandmeyer isatin synthesis method, N-alkylation, condensation, reduction and acid-catalyzed rearrangement. Out of the synthesized compounds 9a–g, compound 9b and 9c exhibited higher cytotoxicity against HeLa cell lines more than other compounds and exhibited cytotoxicity against HepG2 cell lines in similar to compound 9a and 9e but compound 9c is toxic to normal cell lines (Vero). Thus, the substitutions on the isomeric Iheaymine A core structure have to further investigation on their biological activities.
Acknowledgements
We thanks Faculty of Science, Silpakorn University; the Department of Chemistry for the financial support for Rattanopas, S. and the Department of microbiology for the bioactivity assay facilitation and we also thanks the Chulabhorn Research Institute for HREI-MS spectroscopy.
References
- Kohmoto, S.; Kashman, Y.; McConnell, O.J.; Rinehart Jr., K.L.; Wright, A.; Koehn, F. J. Org. Chem. 1988, 53, 3116–3118.
- Moquin-Pattey, C.; Guyot, M. Tetrahedron 1989, 45, 3445–3450.
- Burres, N.S.; Barber, D.A.; Gunasekera, S.P.; Shen, L.L.; Clement, J.J. Biochem. Pharmacol. 1991, 42, 745–751.
- Gunasekera, S.P.; McCarthy, P.J.; Kelly-Borges, M. J. Nat. Prod. 1994, 57, 1437–1441.
- Endo, T.; Tsuda, M.; Fromont, J.; Kobayashi, J. J. Nat. Prod. 2007, 70, 423–424.
- Tanaka, N.; Momose, R.; Takahashi-Nakaguchi, A.; Gonoi, T.; Fromont, J.; Kobayashi, J. Tetrahedron 2014, 70, 832–837.
- Tan, C.-J.; Di, Y.-T.; Wang, Y.-H.; Zhang, Y.; Si, Y.-K.; Zhang, Q.; Gao, S.; Hu, X.-J.; Fang, X.; Li, S.-F.; Hao, X.-J. Org. Lett. 2010, 12, 2370–2373.
- Han, S.; Morrison, K.C.; Hergenrother, P.J.; Movassaghi, M. J. Org. Chem. 2014, 79, 473–486.
- Sasaki, T.; Ohtani, I.I.; Tanaka, J.; Higa, T. Tetrahedron Lett. 1999, 40, 303–306.
- Lindsay, A.C.; Leung, I.K.H.; Sperry, J. Org. Lett. 2016, 18, 5404–5407.
- Abe, T.; Yamada, K. Org. Lett. 2018, 20, 1469–1472.
- Bremner, J.; Sengpracha, W.; Skelton, B. Synthetic Communications. 2008, 38, 1931–1939.
- Teng, Y.-O.; Zhao, H.; Wang, J.; Liu, H.; Gao, M.; Zhou, Y.; Han, K.; Fan, Z.; Zhang, Y.; Sun, H.; Yu, P. Eur. J. Med. Chem. 2016, 112, 145–156.
- Esmaili, A.A.; Bodaghi, A. Tetrahedron 2003, 59, 1169–1171.
- Li, J.; Cheng, X.; Ma, X.; Lv, G.; Zhan, Z.; Guan, M.; Wu, Y. Synlett. 2016, 27, 2485–2488.
- Webber, S.E.; Tikhe, J.; Worland, S.T.; Fuhrman, S.A.; Hendrickson, T.F.; Matthews, D.A.; Love, R.A.; Patick, A.K.; Meador, J.W.; Ferre, R.A.; Brown, E.L.; DeLisle, D.M.; Ford, C.E.; Binford, S.L. J. Med. Chem. 1996, 39, 5072–5082.
- Shmidt, M.S.; Reverdito, A.M.; Kremenchuzky, L.; Perillo, I.A.; Blanco, M.M. Molecules 2008, 13, 831–840.
- Gangar, M.; Kashyap, N.; Kumar, K.; Goyal, S.; Nair, V.A. Tetrahedron Lett. 2015, 56, 7074–7081.
- Kamal, A.; Mahesh, R.; Nayak, V.L.; Babua, K.S.; Kumar, G.B.; Shaik, A.B.; Kapure, J.S.; Alarifi, A. Eur. J. Med. Chem. 2016, 108, 476–485.
- Pei, X.F.; Bi, S. Heterocycles 1994, 39, 357–360.
- Mosmann, T. J. Immunol. Methods 1983, 65, 55–63.
- Hsu, K.-C.; Chen, Y.-F.; Lin, S.-R.; Yang, J.-M. BMC Bioinformatics. 2011, 12, S33.
- Hwang, C.Y.; Lee, S.-M.; Park, S.S.; Kwon, K.-S. Biochem. Biophys. Res. Commun. 2012, 425, 94–99.
- Zalzali, H.; Nasr1, B.; Harajly, M.; Basma, H.; Ghamloush, F.; Ghayad, S.; Ghanem, N.; Evan, G.I.; Saab, R. Mol Cancer Res. 2015, 13, 29–40.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


