Structure-Based Discovery of Inhibitors Against MurE in Methicillin-Resistant Staphylococcus Aureus
Junie B. Billones* and Marielle Alyanna T. Bangalan
and Marielle Alyanna T. Bangalan
Department of Physical Sciences and Mathematics, College of Arts and Sciences University of the Philippines Manila, Padre Faura, Ermita, Manila 1000 Philippines.
Corresponding Author E-mail: jbbillones@up.edu.ph
DOI : http://dx.doi.org/10.13005/ojc/350216
Article Received on : 15-11-2018
Article Accepted on : 13-03-2019
Article Published : 08 Apr 2019
The rise of superbugs is a serious public health concern. It is estimated to kill around 10 million people a year by 2050 and will overtake cancer as the number one cause of death worldwide. One of the most prevalent drug-resistant pathogen is Methicillin-resistant Staphylococcus aureus (MRSA). Intense efforts have been devoted to the discovery and development of anti-MRSA drug. Muramyl ligase E (MurE), an enzyme involved in the peptidoglycan biosynthesis of the bacterial cell wall, is a highly druggable target in MRSA. In this study, virtual screening of approved and experimental drugs in the Drug Bank database was performed based on a pharmacophore derived from the structure of MurE. Molecular docking was subsequently done with the top hits. The top hits and their derivatives were further evaluated for their predicted pharmacokinetics properties. This drug repurposing effort has identified four experimental drugs that are predicted to bind more strongly to MurE than the drug Fosfomycin. One of the top hits, DB01758, exhibited all the characteristics of a good drug candidate, albeit it is likely to be non-biodegradable.
KEYWORDS:Antimicrobial Resistance (AMR); Drug Repurposing; Methicillin-Resistant Staphylococcus Aureus (MRSA); Muramyl Ligase E (Mure); Molecular Docking; Superbug; Virtual Screening
Download this article as:| Copy the following to cite this article: Billones J. B, Bangalan M. A. T. Structure-Based Discovery of Inhibitors Against MurE in Methicillin-Resistant Staphylococcus Aureus. Orient J Chem 2019;35(2). |
| Copy the following to cite this URL: Billones J. B, Bangalan M. A. T. Structure-Based Discovery of Inhibitors Against MurE in Methicillin-Resistant Staphylococcus Aureus. Orient J Chem 2019;35(2). Available from: https://bit.ly/2uPCvQ4 |
Introduction
The drug-resistant microorganisms dubbed superbugs are projected to kill 10 million people a year by 2050 and will overtake cancer as the leading cause of death worldwide.1 The continuous emergence of antimicrobial resistance has resulted to prolonged illnesses, greater treatment costs, and increased mortality making it a public health issue today. The proliferation of superbugs has not only spread in hospital environments but also in communities. The rise of antibiotic resistance has outpaced the development of new antimicrobial drugs in the past 40 years.2 As a result, treatment options for the new strains of these pathogens are severely limited.
In 2014, the World Health Organization (WHO) issued the first ever Antimicrobial Resistance Global Report on Surveillance.3 One of the identified microbial pathogen that has remarkably developed drug resistance is Staphylococcus aureus, a gram-positive and facultative anaerobe. It is one of the leading cause of infection, ranging from minor skin allergies and simple boils to severe lethal conditions such as necrotizing pneumonia and toxic shock syndrome.4,5
Methicillin, like other β-lactam antibiotics such as oxacillin, penicillin, and cephalosporins, was commonly used to treat S. aureus infections.4 Its mechanism involves competitive inhibition of the transpeptidase enzyme used by bacteria to cross-link the D-alanyl-alanine peptide, which is vital for peptidoglycan synthesis.6 Since the discovery of the first methicillin-resistant S. aureus in 1963,7 it has become a primary cause of infection both in hospitals and communities,8 and eventually developed multidrug resistance especially in hospital setting.9
The mechanism of the peptidoglycan biosynthesis is one the best known and most validated drug targets for antibacterial therapy.10,11 The compounds that interfere with the biosynthesis and assembly of peptidoglycan have been utilized as antimicrobial agents. This makes the Mur enzymes, which catalyze the cytoplasmic steps of the peptidoglycan biosynthesis, excellent candidates for drug development.12 The high tensile strength and rigidity of the bacterial cell wall is attributed to the cross-linkage of linear glycan chains which are composed of alternating units of N-acetylglucosamine and N-acetylmuramic acid.13
In the cytoplasm, MurA catalyzes the acylation of UDP-N-acetylglucosamine by phosphoenol pyruvate. The subsequent reduction of the product enol-pyruvoyl-UDP-N-acetylglucosamine with NADPH is catalyzed by MurB. The resulting product UDP-N-acetylmuramic acid (UDP- MurNAc) is then aminoacylated, in sequence, with L-alanine, D-glutamate, and either L-lysine or meso-diaminopimelic acid (mDAP) by MurC, MurD, and MurE, respectively, to produce UDP-MurNAc-tripeptide. Finally, the prostaglandin precursor UDP-MurNAc-pentapeptide is produced by MurF-catalyzed addition of D-alanyl-D-alanine to the carboxyl end of the UDP-MurNAc-tripeptide.14 The pentapeptides in prostaglandin strands are cross-linked by transpeptidation catalyzed by penicillin-binding proteins (PBPs), which link the MurE appended L-lysine or mDAP residue to adjacent peptides.15 This amino acid in third position is an important site for elaboration of the stem peptide in prostaglandin and serves as linchpin for diverse cell wall biomolecules, with the aid of sortase enzymes that are crucial for virulence and pathogenesis.16 It is therefore imperative to find inhibitors of MurE enzyme, which is essential in the crucial step of prostaglandin biosynthesis.
In this work, pharmacophore-based virtual screening was performed with the use of DrugBank database (www.drugbank.ca) containing around 7000 compounds, including over 2000 approved drugs and some 5000 experimental drugs and nutraceuticals. Subsequently, the top hits were subjected to molecular docking studies against MurE as enzyme target. The high-binding top hits were further evaluated for their pharmacokinetics properties.
Material and Methods
All computational works were performed using Biovia Discovery Studio (DS) Client v2.5 (Dassault Systèmes) installed on a computer running on Microsoft Windows 7 Professional 64-bit Operating System using a 3.50-GHz Intel® Core™ i7-4770K processor with 8.00 GB memory (RAM). The crystallographic data file of the target, MurE (PDB ID: 4C13), was retrieved from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) website (www.rcsb.org).
After removing the co-crystallized ligand, the enzyme was defined as the receptor using the Define Selected Molecule as Receptor tool in DS. The location of the bound ligand was used to generate an active site sphere using Define Sphere from Selection tool. The nature of the active site of the target molecule was analyzed for hydrogen bonding and hydrophobic features using Interaction Generation protocol. The initial pharmacophore features were narrowed down using the Cluster Current Feature and Keep Only Cluster Centers tools for the generated features.
The SDF files of the comprehensive online database of drugs were obtained from DrugBank. Using the Prepare Ligands protocol, the compounds were prepared by removing duplicates, varying the ionization based on pH (6.5 to 8.5), enumerating tautomers and isomers, and generating 3D conformations. The prepared ligands were assembled into a separate database using the Build 3D Database protocol while the number of conformation was set to 0.
The prepared database was screened against the pharmacophore generated using the Screen Library protocol by rigid fitting method. The compounds that displayed fit values of at least 3.8 were subjected to molecular docking studies using the Dock Ligands (CDOCKER) protocol. The target-ligand complexes were evaluated and ranked by calculating their binding energies using the Calculate Binding Energies protocol with In Situ Ligand Minimization.
The toxicity and ADMET (absorption, distribution, metabolism, excretion, toxicity) characteristics were determined using the TOPKAT and ADMET modules in DS.
Results and Discussion
The target enzyme MurE ligase from S. aureus (MurESa) in complex with UDP-MurNAc-Ala-Glu-Lys was retrieved from RCSB Protein Data Bank. It was subjected to Protein Preparation step which involves insertion of missing atoms and loop regions, deletion of alternate conformations, removal of water and ligands, and protonation of amino acid residues based on pKa values. Additionally, the energy of the prepared structure was minimized through geometry optimization, thereby obtaining the most stable conformation of the protein. The optimized structure of the target did not vary considerably vis-à-vis the original crystal structure after protein preparation (RMSD = 0.91Å). The 3D structure of MurESa prepared in DS was exported to and visualized as ribbon representation (chain A) in UCSF Chimera v 1.11.217 (Figure 1). MurESa is a three-domain, mixed a/b structure. Domain 1 includes residues 1 to 98, which comprise the uridine nucleoside-binding site of the UDP-MurNAc-tripeptide product. Domain 2 covers residues 99 to 332 and encompasses the rest of the UDP-MurNAc-tripeptide-binding pocket. Lastly, domain 3 embraces residues 333 to 493 and partly encompasses the ATP-binding site.10 The inset displays the hydrophobicity surface of the enzyme around the UDP-MurNAc-tripeptide-binding pocket. The tripeptide moiety appears to extend down to the buried ATP-binding site formed between domain 2 and 3.
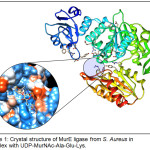 |
Figure 1: Crystal structure of MurE ligase from S. Aureus in complex with UDP-MurNAc-Ala-Glu-Lys. |
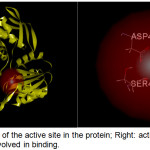 |
Figure 2: Left: Location of the active site in the protein; Right: active site containing amino acid residues involved in binding. |
A site sphere with a radius of 7.5 Å and x, y and z coordinates of -29.5, 8.5, and -3.0 respectively was positioned around domains 2 and 3 in accord with the active site previously described by Ruane and coworkers [10]. The site sphere radius was optimized by varying its size and position until all relevant amino acid residues in the binding site, specifically Glu-460, Asp-406, and Ser-456) were covered (Figure 2). COACH and COFACTOR servers (https://zhanglab.ccmb.med.umich.edu) were also utilized to check the active site of the target enzyme selected. These servers provide complementary ligand binding site predictions via two comparative methods, TM-SITE and S-SITE.18 These methods recognize ligand-binding templates from the BioLiP protein function database19 by binding-specific substructure and sequence profile comparisons.
A pharmacophore, a set of steric and electronic features of a compound needed to ensure optimal interactions with a specific biological structure, was then generated based on the structure of the defined binding pocket. Figure 3 shows that it consists of 31 features: 15 acceptors, 14 donors and 2 hydrophobes. The different colored spheres correspond to the spatial arrangement of different features – green for hydrogen-donors, magenta for hydrogen-acceptors, and cyan for the hydrophobes. These features served as reference in screening the DrugBank database of approved and experimental drugs.
Using the Screen Library protocol, structure-based virtual screening was performed in order to reduce the number of compounds to be docked against the target. Applying an arbitrary cut-off fit value of 3.8, the number of experimental compounds was trimmed down from 4,982 to 346 and the approved drugs from 2,162 to 947.
 |
Figure 3: Generated pharmacophore having 31 features: 15 acceptors (magenta), 14 donors (green) and 2 hydrophobes (cyan). |
Molecular docking is an established tool in molecular biology and drug discovery.20 It has been routinely used in our group to perform virtual screening of libraries and to elucidate the interaction of the ligands with a biomolecular target.21-27 Thus, molecular docking was subsequently performed in order to rank-order the pharmacophore-based virtual screening hits according to binding energy values and to examine the interaction of the ligands against the target. Using the CDOCKER protocol in DS,28 which employs a CHARMm (Chemistry at HARvard Macromolecular Mechanics) force field, random poses of a ligand were generated and placed in a rigid receptor (i.e. defined binding pocket in MurESa). The resultant poses were then evaluated by calculating their binding energies.
The ATP-binding site in MurESa has been established29 and is composed of structural features that are conserved in all Mur ligases [10]. In this light, Fosfomycin, a broad-spectrum antibiotic which acts by inactivating MurA, was used as reference in this work. To date, there is no known drug that acts against MurE and so far its only known inhibitor is a phosphinate.30 Thus, the screened ligands with a more negative binding energy than Fosfomycin (-335.40 kJ/mol) were considered as promising top hits.
Molecular docking studies yielded four compounds (Table 1) with greater (more negative) binding energy than the reference compound. The topmost hit was an experimental compound, 4-Phospho-L-Threonic acid (DB01756), a monosaccharide phosphate that acts on ribose-5-phosphate isomerase. Its binding affinity is relatively more than double that of Fosfomycin. The second top hit was an inositol phosphate, (1S,3S,4S)-1,3,4-Triphospho-Myo-Inositol (DB01729), which is also an experimental drug. Its binding energy with MurESa is more than 100 kcal/mol greater than that of the reference compound. The third most potent hit was 3-Iodo-Tyrosine (DB01758), an organic acid derivative that targets tyrosine-tRNA ligase. It is the only hit compound that does not contain a phosphate group. Lastly, the fourth hit was N-Hydroxy-4-Phosphono-Butanamide (DB01695), an experimental compound that targets triosephosphate isomerase.
The top hit compound, DB01756, was found to have more polar, van der Waals, and H-bonding interactions with MurESa compared to Fosfomycin. DB01756 exhibits hydrogen bonding with the side chains of Thr137, Gly149, Thr152, and Tyr 462 (Figure 4); charge interaction with Lys148; and several polar interactions with Asn138, Ala150, Asn151, Tyr351, Gly460, and Tyr 462 (Figure 5a). The ligand-target interaction maps for the other top hit molecules are also displayed in Figure 5.
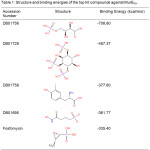 |
Table 1: Structure and binding energies of the top hit compounds against MurESa. |
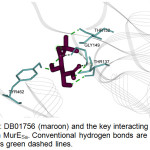 |
Figure 4: DB01756 (maroon) and the key interacting residues (cyan) in MurESa. Conventional hydrogen bonds are shown as green dashed lines. |
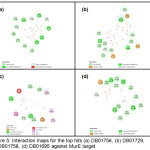 |
Figure 5: Interaction maps for the top hits (a) DB01756, (b) DB01729, (c) DB01758, (d) DB01695 against MurE target. |
Table 2: Predicted Pharmacokinetics Properties of the Top Hit Compounds.
|
Compound |
Human Intestinal Absorptiona |
Aqueous Solubilityb |
CYP2D6 Bindingc |
Hepatotoxicityd |
Plasma Protein Bindinge |
|
DB01756 |
3 |
5 |
0 |
0 |
0 |
|
DB01729 |
3 |
4 |
0 |
0 |
0 |
|
DB01758 |
0 |
4 |
0 |
0 |
0 |
|
DB01695 |
3 |
5 |
0 |
0 |
0 |
|
Fosfomycin |
2 |
5 |
0 |
0 |
0 |
a 0 = good absorption, 1 = moderate absorption, 2 = poor absorption, 3 = very poor absorption; b 0 = low; 1 = no, very low, but possible; 2 = yes, low; 3 = yes, good; 4 = yes, optimal; 5 = too soluble; c 0 = non-inhibitor, 1 = inhibitor; d 0 = non-toxic, 1 = toxic; e 0 = < 90% (not highly bound), 1 = > 90% (highly bound), 2 = > 95% (highly bound).
The ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) properties of the top hits were also predicted using the ADMET protocols in DS. The Human Intestinal Absorption protocol was used to predict the human intestinal absorption after oral administration and ingestion. The Aqueous Solubility protocol predicts the solubility of each compound in water at 25°C. ADMET CYP2D6 Binding predicts cytochrome P450 2D6 enzyme inhibition and classifies whether a compound is likely to be an inhibitor or not. Hepatotoxicity predicts the occurrence of dose-dependent human hepatotoxicity. The Plasma Protein Binding model predicts whether a compound is likely to be bound to carrier proteins present in the blood. Theoretically, a good drug exhibits the following characteristics: good human intestinal absorption, optimal aqueous solubility, non-CYP2D6 inhibitor, low hepatotoxicity, and is not highly-bound. Among the top hits DB01758 has the all the characteristics of a good drug based on ADMET parameters. Although DB01756 has the largest binding energy, it is too soluble in water and not easily absorbed by the intestine. With the exception of DB01758, the top hits including the drug Fosfomycin are predicted to have poor to very poor intestinal absorption. However, they can be alternatively delivered to the target using an appropriate drug delivery system.31
The TOxicity Prediction by Komputer Assisted Technology (TOPKAT) protocols in DS28 were used to predict the toxicity properties of the top hit compounds. TOPKAT provides assessment of chemical toxicity based on QSAR models that involve predictors derived solely from chemical structure. Based from the Weight-of-Evidence Rodent Carcinogenicity module of TOPKAT, a probability of 0.3 and below indicates that the compound is non-carcinogenic while a probability above 0.7 entails that the compound is carcinogenic.32 The Ames Mutagenicity module, on the other hand, labels a compound as non-mutagenic if the computed mutagenicity is between 0.0 and 0.3. The Developmental Toxicity module aids in determining whether or not a drug may be administered to pregnant women or children by predicting the toxicity of a drug candidate to the embryo or fetus. Finally, the Aerobic Biodegradability module predicts the stability of the compound by calculating the probability of a query to be capable of aerobic biodegradation.28 A good drug exhibits the following properties: non-carcinogenic, non-mutagenic, exhibits no developmental toxicity potential, and is biodegradable. Among the top hit compounds, DB01758 was predicted to have all the characteristics of a good drug, except that it is non-biodegradable (Table 3). In general, the calculated ADME-Tox properties obtained from this work are in agreement with the admet SAR-predicted properties listed in Drug Bank.
Table 3: Predicted Toxicity Properties of the Top Hit Compounds.
|
Compound |
Carcinogenicity |
Mutagenicity |
DevelopmentalToxicity Potential |
Aerobic Biodegradability |
|
DB01756 |
non-carcinogenic |
mutagenic |
– |
biodegradable |
|
DB01729 |
carcinogenic |
non-mutagenic |
+ |
biodegradable |
|
DB01758 |
non-carcinogenic |
non-mutagenic |
– |
non-biodegradable |
|
DB01695 |
non-carcinogenic |
non-mutagenic |
– |
biodegradable |
|
Fosfomycin |
non-carcinogenic |
non-mutagenic |
– |
non-biodegradable |
Conclusion
In this study, a pharmacophore based on the structure of MurESa was generated and used to screen a total of 7,144 approved and experimental drugs from Drug Bank database. The top hits were then docked to the enzyme target and their binding energies were calculated. Four compounds displayed greater inhibitory potential against MurE ligase compared to Fosfomycin, a drug known to act on structurally related MurA ligase. Three of the top hits possess a phosphate group and complements the defined binding site in MurE, which encompasses an ATP-binding pocket that sits between domains 2 and 3 of the enzyme. The topmost hit was an experimental drug, 4-Phospho-L-Threonic acid (DB01756), which is a monosaccharide phosphate that reportedly acts on ribose-5-phosphate isomerase. Interestingly, 3-Iodo-Tyrosine (DB01758), the only hit compound that does not contain a phosphate group, possesses the desirable properties of a drug-like molecule. The experimental drugs identified in this drug-repositioning effort can be further explored for their inhibitory activity against MurE ligase, which is essential in bacterial cell wall biosynthesis.
Acknowledgements
This work is supported by the Office of the Vice President for Academic Affairs (OVPAA), University of the Philippines System through the Enhanced Creative Work and Research Grant (ECWRG 2017-1-032).
References
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations, The Review on Antimicrobial Resistance, 2016. Retrieved from https://amr-review.org.
- Spellberg, B.; Powers, J.H.; Brass, E.P.; Miller, L.G.; Edwards, J.E., Jr. Clin. Infect. Dis., 2004, 38: 1279 – 1286.
- World Health Organization (WHO). Antimicrobial Resistance: global report on surveillance 2014. Retrieved from http://www.who.int.
- Patnala, K.; Zaveri, K. Asian J. Pharm. Clin. Res., 2016, 283.
- Pardeshi, K.A.; Malwal, S.R.; Banerjee, A.; Lahiri, S.; Rangarajan, R.; Chakrapani, H. Bioorg. Med. Chem. Lett., 2015, 25(13), 2694 – 2697.
- Mitscher, L. Antibiotics and antimicrobial agents. In: Williams, D.A.; Lemke, T.L., Eds. Foye’s principles of medicinal chemistry. Philadelphia: Lippincott Williams & Wilkins, 2002.
- Jevons, M.P.; Coe, A.W.; Parker, M.T. Lancet, 1963, 27; 1(7287): 904 – 907.
- Huang, H.; Flynn, N.M.; King, J.H.; Monchaud, C.; Morita, M.; Cohen, S.H. J. Clin. Microbiol. 2006, 44(7): 2423 – 2427.
- Boucher, H.W.; Corey, G.R. Clin. Infect. Dis. 2008, 46 Suppl 5: S344 – 349.
- Ruane, K.; Lloyd, A.; Fülöp, V.; Dowson, C.; Barreteau, H.; Boniface, A. et al. J. Biol. Chem., 2013, 288(46), 33439 – 33448.
- Tomašić, T.; Šink, R.; Zidar, N.; Fic, A.; Contreras-Martel, C.; Dessen, A.; … Mašič, L.P. ACS Med. Chem. Lett., 2012, 3(8), 626 – 630.
- Nikolaidis, I.; Favini-Stabile, S.; Dessen, A. Protein Sci., 2014, 23(3), 243 – 259.
- Laddomada, F.; Miyachiro, M.; Dessen, A. Antibiotics, 2016, 5(2): 14.
- Barreteau, H.; Kovac, A.; Boniface, A.; Sova, M.; Gobec, S.; Blanot, D. FEMS Microbiol. Rev., 2008, 32: 168 – 207.
- Matteï, P.J.; Neves, D.; Dessen, A. Curr. Opin. Struct. Biol., 2010, 20: 749 – 755.
- Clancy, K.W.; Melvin, J.A.; McCafferty, D.G. Biopolymers, 2010, 94: 385 – 396.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. J. Comput. Chem., 2004, 25(13): 1605 – 1612.
- Yang, J.; Roy, A.; Zhang, Y. Bioinformatics, 2013, 29: 2588 – 2595.
- Yang, J.; Roy, A.; Zhang, Y. Nucleic Acids Res., 2013, 41: D1096 – D1103.
- Morris, G.M.; Lim-Wilby, M. Methods Mol. Biol., 2008, 443: 365 – 382.
- Billones, J. B.; Carrillo, M. C. O.; Organo, V. G.; Sy, J. B.; Clavio, N. A. B.; Macalino S. J. Y.; Emnacen, I. A.; Lee, A. P.; Ko, P. K. L.; Concepcion, G. P.; Drug Des. Devel. Ther., 2017, 11, 563 – 574.
- Billones, J.B.; Carrillo, M. C. O.; Organo, V.G.; Macalino, S. J.; Sy, J. B.; Emnacen, I. A.; Clavio, N. A.; Concepcion, G. P. Drug Des. Devel. Ther., 2016, 10, 1147 – 1157.
- Sampaco III, A. B.; Billones, J. B. Orient J. Chem., 2015, 31(4), 1859 – 1865.
- Billones, J. B.; Carrillo, M. C. O.; Organo, V. G.; Macalino, S. J. Y.; Emnacen, I. A.; Sy, J. B. Curr. Enzym. Inhib., 2014, 10(2), 105 – 112.
- Billones, J. B.; Valle, A. M. F. Orient. J. Chem., 2014, 30(3), 1137 – 1145.
- Billones. J. B.; Carrillo, M. C. O.; Organo, V. G. Macalino, S. J. Y.; Emnacen, I. A.; Sy, J. B. A. Orient. J. Chem., 2013, 29(4), 1457 – 1468.
- Billones, J. B. Orient. J. Chem., 2016, 32(2), 851 – 858.
- Accelrys Software Inc. Help Topics (2014).
- Walker, J. E.; Saraste, M.; Runswick, M.J.; Gay, N.J. EMBO J. 1982, 1: 945 – 951.
- Zeng, B.; Wong, K.; Pompliano, D.; Reddy, S.; Tanner, M. J. Org. Chem., 1998, 63: 10081 – 10086.
- Mishra, M. Degradation of polymers. Handbook of encapsulation and controlled release. Taylor & Francis Group. 2016, 584 – 585.
- Worth, A.; Lapenna, S.; Lo, P.; Mostrag-Szlichtyng, A.; Serafimova, R. The Applicability of Software Tools for Genotoxicity and Carcinogenicity Prediction: Case Studies relevant to the Assessment of Pesticides. Publications Office of the European Union. 2010. Retrieved from http://doi.org/10.2788/61485.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


