Retusenol Potentially Inhibits Putative Drug Targets for Tuberculosis, Cardiovascular Diseases, Cancer and HIV: A Reverse Docking Study
Peñafrancia D. Rariza and Junie B. Billones
Department of Physical Sciences and Mathematics, College of Arts and Sciences University of the Philippines Manila, Padre Faura, Ermita, Manila 1000 Philippines.
Corresponding Author E-mail: jbbillones@up.edu.ph
DOI : http://dx.doi.org/10.13005/ojc/3404012
Article Received on : 20-05-2018
Article Accepted on : 24-07-2018
Article Published : 21 Aug 2018
Retusenol is a new triterpene discovered from the leaf extracts of Atalantia retusa Merr., a Philippine endemic shrub. With the emergence of drug resistance and drug tolerance in certain pathological conditions, it is a worthwhile endeavor to find possible medicinal applications of this natural product. Molecular docking of retusenol was performed against proteins that are putative drug targets in diverse diseases. Four enzymes were found to exhibit stronger binding affinity to Retusenol compared to their known inhibitors, namely, enoyl-acp reductase, lanosterol synthase, polo-like kinase 1, and integrase. These enzymes are drug targets for tuberculosis, cardiovascular diseases, cancer and HIV, respectively.
KEYWORDS:Enoyl-acp reductase; Integrase, Lanosterol synthase; Polo-like kinase 1; Retusenol; Reverse Docking
Download this article as:| Copy the following to cite this article: Rariza P. D, Billones J. B. Retusenol Potentially Inhibits Putative Drug Targets for Tuberculosis, Cardiovascular Diseases, Cancer and HIV: A Reverse Docking Study. Orient J Chem 2018;34(4). |
| Copy the following to cite this URL: Rariza P. D, Billones J. B. Retusenol Potentially Inhibits Putative Drug Targets for Tuberculosis, Cardiovascular Diseases, Cancer and HIV: A Reverse Docking Study. Orient J Chem 2018;34(4). Available from: http://www.orientjchem.org/?p=48713 |
Introduction
Retusenol was isolated from Atalantia retusa Merr., an endemic shrub of Mindoro, Palawan and Panay islands, Philippines. It demonstrated anti-microbial property against five microorganisms, namely: B. subtilis, P. aerugininosa, S. aureus, C. albicans, and T. mentagrophytes [1]. Retusenol ((2S,4aS,6aR,7S,8S,10aS,10bS,12aR)-1,1,4a,6a,8,10a-hexamethyl-7-(4-methylpentyl)-1,2,3,4,4a,6,6a,7,8,9,10,10a,10b,11,12,12a-hexadecahydrochrysen-2-ol), a triterpene, was found to be a colorless solid with a melting point of 145°C.1
Natural products have served as the source and inspiration for a greater portion of the current pharmacopoeia. It is estimated that between 25% and 50% of currently marketed drugs owe their origins to natural products.2 Thus, it is certainly worthwhile to find therapeutic applications of the newly isolated natural products like Retusenol considering the ever growing cases of drug resistance and drug tolerance. Drug resistance happens when microorganisms such as bacteria, viruses, fungi and parasites respond in such a way that the drug is rendered ineffective as cure for the infections. It has become a serious health problem because a resistant infection may cause death, can spread to others, and incurs huge costs to individuals and society.3 On the other hand, drug tolerance is a decrease in response to a drug dose that occurs with repeated use.4 Indeed, there is a serious need to develop new drugs in lieu of existing drugs that are rendered ineffective due to drug resistance or drug tolerance.
Computer-based technologies have been employed in drug discovery efforts today in order to speed up the intricate stages in drug discovery. In particular, molecular docking has been commonly applied in screening large molecular databases against a putative druggable macromolecular target, rank order the results, and predict the nature of the ligand-target interaction, which is crucial in lead optimization.5 Conversely, it can also be applied in screening a database of druggable targets against certain drug-like molecules,6 a technique called reverse or inverse docking.7,8 In structure-based drug design, in which the structure of the drug target is known, molecular docking is one of the most frequently used computational methods due to its ability to predict the binding conformation of ligands to the binding site.9 This well-established technique enables the characterization of the binding behavior which is crucial in the rational design of drugs and in the elucidation of fundamental biochemical processes. Our group has routinely utilized both normal molecular docking10-20 and inverse docking techniques21 in our drug discovery efforts.
In this work, Retusenol was docked to a number of enzymes that are associated with different pathological states and are considered highly druggable targets. The identification of proteins that strongly interact with Retusenol will indicate possible utility of the title compound in the treatment of the corresponding disease.
Materials and Methods
Molecular docking studies were done with the use of Discovery Studio 2.5 (DS; BIOVIA-Dassault Systèmes, formerly Accelrys) running on Windows 7 operating system in a machine with an Intel® CoreTM i7-3770 and 3.40GHz quad core processor. Spartan 16 (Wavefunction, Inc.) was used to model the 3D structure of the Retusenol ligand. All enzymes and inhibitors were retrieved from RCSB Protein Data Bank (http://www.rcsb.org). The protein structures of the enzymes were cleaned using the Clean Protein Protocol of DS. Additionally, some corrections were done in preparing the protein, such as removal of alternate conformations, addition of hydrogen to hetero atoms and correcting for missing or incorrectly specified residues. All unnecessary substances such as ligands and cofactors were removed from the protein model. The prepared protein was defined as the receptor using the Define Selected Molecule as Receptor tool. The site spheres were chosen using the Define Sphere from Selection Tool. The site spheres were expanded to a radius of more than 10 Å. The prepared ligands and proteins were docked using the Dock Ligands (CDOCKER) protocol of DS. The Calculate Binding Energies protocol employing CHARMm forcefield was used to calculate binding energies for the proteins and the docked ligands. The Draw Ligand Interaction Diagram Tool was used to determine and visualize the non-covalent interactions between the protein and the docked ligands.
Results and Discussion
Retusenol was chosen as the target ligand because it is a relatively new natural product, thus it is still largely unexplored and its uses are practically unknown apart from its moderate antimicrobial activity.1 In addition, the investigators have special interest on this compound considering that it was discovered from an endemic plant in their country. Interestingly, it exhibited anti-microbial properties against five microorganisms. Retusenol is a tetracyclic compound with only alcohol and alkene functionalities (Figure 1). Figure 1b shows that the molecule is largely hydrophobic and the only polar part is the ring that bears the lone hydroxyl group, as indicated by the negative electrostatic potential (red at lower left side) on the isosurface of the molecule.
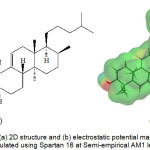 |
Figure 1: The (a) 2D structure and (b) electrostatic potential map of Retusenol calculated using Spartan 16 at Semi-empirical AM1 level. |
Figure 2 shows an example of a prepared protein, particularly Lanosterol synthase with a defined binding site. In molecular docking using the CDOCKER protocol, different ligand conformations were obtained using high-temperature molecular dynamics, then random orientations of the conformations were generated by moving the center of the ligand to a particular position within the receptor active site, and a series of random rotations were performed. An orientation was kept if the energy was less than a specified threshold. The poses were sorted by CHARMm energy and the top scoring (most negative value) poses were retained. Figure 3 shows an example of a successfully docked ligand, i.e. Retusenol to the prepared protein Lanosterol synthase.
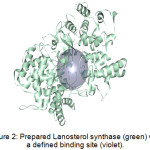 |
Figure 2: Prepared Lanosterol synthase (green) with a defined binding site (violet). |
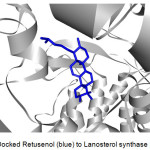 |
Figure 3: Docked Retusenol (blue) to Lanosterol synthase (gray). |
The enzymes that exhibited successful docking with Retusenol were further studied. That is, the known ligands of these enzymes were determined and also docked to the corresponding enzyme as exemplified by the docking of Lanosterol, a known ligand of Lanosterol synthase (Figure 4).
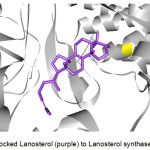 |
Figure 4: Docked Lanosterol (purple) to Lanosterol synthase (gray). |
Binding energy is the Gibbs free energy associated with the formation of ligand-target complex from its constituent parts. It is the sum of the free energy contributions of non-covalent interactions (H-bonding, electrostatic, hydrophobic, etc). A more negative binding energy indicates that the formation of the complex is more favored thermodynamically and indicates stronger binding interaction, which can be translated as greater inhibitory potential (i.e. potency). To illustrate, a molecule with a 10 nM binding constant roughly corresponds to an overall Gibbs free binding energy of 46 kJ mol−1. At 300 K, a 10-fold increase in potency was noted for every ∼5.7 kJ mol−1 gain in binding energy.22
The calculated binding energies of Retusenol with target enzymes are shown in Table 1. For comparison, the binding energies of known ligands of the enzymes are also listed. Among the enzymes that yielded a negative binding energy with Retusenol, four were calculated to have a greater (more negative) binding energy compared to the binding energy with its known inhibitor. These enzymes were the following: Enoyl-acp reductase, Lanosterol synthase, Polo-like kinase 1, and Integrase. Enoyl-acp reductase, Lanosterol synthase, Polo-like kinase 1 and Integrase are drug targets for Tuberculosis, Cardiovascular diseases, Cancer, and HIV, respectively.
Table 1: Calculated Binding Energies of Retusenol and known inhibitors to different enzymes.
|
Enzyme |
Known Ligand/Inhibitor |
Binding Energy (kcal/mol) |
|
| Retusenol | Inhibitor | ||
| Alpha-glucosidase | Maltose |
-47.66 |
-130.95 |
| Beta secretase | Spiropiperdine iminohydantoin |
-66.86 |
-116.74 |
| Cytochrome P450 3a4 | Erythromycin |
-118.00 |
-146.03 |
| Dextran glucosidase | Isomaltotriose |
-78.25 |
-188.22 |
| Dihydrofolate reductase | Methotrexate |
-72.32 |
-449.88 |
| Dipeptidyl peptidase 4 | Sitagliptin |
-47.31 |
-279.75 |
| Enoyl-acp reductase | Isoniazid |
-44.47 |
-30.69 |
| Estrogen receptor alpha | Tamoxifen |
-33.92 |
-171.69 |
| Glucoamylase | Acarbose |
-30.51 |
-195.42 |
| Heat shock protein 90 | Geldanamycin |
-58.97 |
-144.93 |
| HIV-1 protease | Amprenavir |
-80.60 |
-127.14 |
| Hmg-coa reductase | Atorvastatin |
-108.06 |
-290.04 |
| Human chymase | Benzimidazolone |
-20.9 |
-192.80 |
| Human mdm2 | Dihydroimidazothiazole |
-54.38 |
-69.44 |
| Integrase | Elvitegravir |
-26.44 |
-13.19 |
| Lanosterol synthase | Lanosterol |
-46.30 |
-35.77 |
| Neuraminidase n2 | Sialic acid |
-61.32 |
-180.93 |
| Polo-like kinase 1 | Pyrazoloquinazoline |
-77.96 |
-75.34 |
| Proto-oncogene tyrosine-protein kinase src | Quinazoline |
-59.98 |
-125.95 |
| Serine/threonine-protein kinase 6 | Imidazopyrazine |
-44.54 |
-89.62 |
| Tie2 kinase | Thiazolopyrimidine |
-56.26 |
-67.46 |
Enoyl–acp reductase is essential in fatty acid biosynthesis, which is crucial for cell viability.23 It is the enzyme that promotes the hydrogenation of acyl-ACP complexes to trans-2,3-dehydroacyl-ACPs, with NADP+ as hydrogen acceptor.24 It catalyzes the last step of fatty acid elongation. Enoyl–acp reductase has become an attractive target for the development of antibacterial agents.25 The binding affinity of Retusenol with enoyl-acp reductase was about 15 kcal/mol stronger than that of Izoniazid, a first line anti-TB drug and a known inhibitor of the enzyme, although it needs activation by binding with NADH.26
The enzyme, Lanosterol synthase, is important in the inhibition of cholesterol biosynthesis.27 The conversion of (S)-2,3-oxidosqualene to lanosterol, a four-ring sterol intermediate, is a key step in the biosynthesis of cholesterol and steroid hormones.28 Identification of drugs that targets this enzyme for the treatment of hypercholesterolemia is of great interest. The binding energy of Retusenol in complex with lanosterol synthase is about 10 kcal/mol stronger than that of Lanosterol.
Serine/Threonine-protein kinase 1 or Polo-like kinase 1 (PLK1) is highly expressed in many cancers and regulates crucial steps in mitotic progression.29,30 The oncogenic properties of PLK1 are attributed to its role in driving cell cycle progression as evidenced by its overexpression in NIH3T3 cell line.31 Inhibition of Polo-like kinase 1 decreased tumor growth both in vitro and in vivo. Retusenol slightly outperformed Pyrazoloquinazoline, a known inhibitor of polo-like kinase 1.
Integrase is the enzyme that splices the viral DNA into a host’s cellular chromosome.32 Specifically, it prepares an area on the viral DNA for integration, and then transfers the processed strand into the genome of the host cell. Thus, integrase is essential for viral replication. With integrase as target, the strength of Retusenol binding is twice that of its known ligand Elvitegravir.
Additionally, the docking of Retusenol to three targets, namely, Glycogen phosphorylase b, Cytochrome p450 19a1, and Serine/threonine – protein kinase 12-a were also successful, although the binding energy values were all positive and more positive than that with the corresponding known inhibitor. Furthermore, Retusenol failed to dock to several other enzymes using the same docking parameters as above. These enzymes were the following: Acetylcholinesterase, Angiotensin converting enzyme, Kinesin-like protein kif11, Thymidine kinase, Malarial purine phosphoribosyl transferase, Prostaglandin synthase-1 and Prostaglandin synthase-2. The active sites of these targets are probably not large enough to accommodate Retusenol, a tetracyclic compound.
The type and number of interactions made by Retusenol with its top four targets are detailed in Table 2 and illustrated in Figure 3 (with Lanosterol synthase) and Figure 5 [a) with enoyl-acp reductase, b) polo-like kinase 1, and c) integrase]. The non-covalent interactions of the known inhibitor of each target are also listed for comparison. The binding affinity of Retusenol with enoyl-acp reductase and polo-like kinase 1 was almost exclusively van der Waals in nature. With the much smaller ligand Izoniazid, the interaction with enoyl-acp reductase was a combination of two H-bonds, 4 van der Waals, and 6 polar interactions. Pyrazoloquinazoline exhibited fewer van der Waals interaction with polo-like kinase 1, albeit it featured few H-bonds, pi, and polar interactions. Retusenol outperforms the substrate of lanosterol synthase because of the extra polar interaction. Although Elvitegravir showed more interactions with integrase compared to Retusenol, the strength of these interactions and the presence of unfavorable ones explain the observed discrepancy in binding energies.
Table 2: Type and number of Interactions of Retusenol and Known Inhibitors of Hit Enzymes
| Enzymes |
Compound |
Interactions |
||||
| Charged | H-bonding | Van der Waals | Pi | Polar | ||
| Enoyl-acp reductase | Retusenol |
0 |
0 |
19 |
0 |
0 |
| Isoniazid |
0 |
2 |
4 |
0 |
6 |
|
| Lanosterol synthase | Retusenol |
1 |
26 |
1 |
3 |
|
| Lanosterol |
0 |
1 |
26 |
0 |
2 |
|
| Polo-like kinase 1 | Retusenol |
0 |
0 |
16 |
0 |
1 |
| Pyrazoloquinazoline |
0 |
3 |
13 |
3 |
7 |
|
| Integrase | Retusenol |
0 |
0 |
4 |
0 |
1 |
| Elvitegravir |
0 |
0 |
6 |
0 |
2 |
|
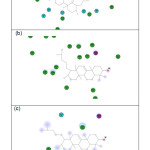 |
Figure 5: Interaction diagram for Retusenol and (a) Enoyl-acp reductase, (b) Polo-like kinase 1, and (c) Integrase. Click here to View figure |
Residues involved in hydrogen-bonding, charge or polar interactions are represented by magenta-colored circles, those involved in van der Waals interactions are represented by green circles, water molecules are represented by aquamarine circles, the solvent accessible surface of an interacting residue is represented by a blue halo around the residue. The diameter of the circle is proportional to the solvent accessible surface.
Conclusion
Retusenol, a new triterpene from a Philippine endemic plant named Atalantia retusa Merr. was molecularly docked with enzymes that are putative drug targets for different diseases. Among the enzymes studied, four were found to have stronger binding affinity with Retusenol than to their corresponding known ligand. These enzymes are the following: Enoyl-acp reductase, Lanosterol synthase, Polo-like kinase 1, and Integrase. These enzymes are.
Acknowledgement
The authors are grateful to the Emerging Inter-Disciplinary Research (EIDR) program (OVPAA-EIDR 12-001- 121102) for the use of its computational facilities.
drug targets for the treatment of Tuberculosis, Cardiovascular diseases, Cancer and HIV, respectively. The results in this computational study indicate that the compounds based on Retusenol chemical architecture may be strongly pursued as drug leads for the aforementioned diseases.
References
- Ragasa, C.Y.; Espineli, D. L.; Mandia, E. H.; Raga, D. D.; Don, M.; Shen, C. Z. Naturforsch. 2012, 67b, 426 – 432
CrossRef - Kingston, D.G. J. Nat. Prod, 2011, 74, 496 – 511.
CrossRef - WHO. Antimicrobial Resistance, n.d.. (Retrieved from http://www.who.int/topics/drug_resistance/en/).
- National Institute on Drug Abuse (NIDA), The Neurobiology of Drug Addiction, 2017. (Retrieved from https://www.drugabuse.gov/neurobiology-drug-addiction).
- Morris, G. M.; Lim-Wilbey, M. Methods Mol. Biol. 2008, 443, 365 – 382
CrossRef - Zheng, R.; Chen, T.; Lu, T. Int. J. Mol. Sci. 2011,12(8), 5200 – 5212
CrossRef - Chen, Y. Z.; Ung C.Y. J. Mol. Graph. Model. 2001, 20(3), 199 – 218
CrossRef - Li, H.; Gao, Z.; Kang, L.; Zhang, H.; Yang, K.; Yu, K.; Luo, X.; Zhu, W.; Chen, K.; Shen, J.; Wang, X.; Jiang, H. Nucleic Acids Res. 2006, 34, W219 – 224
CrossRef - Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Nat. Rev. Drug Discov. 2004, 3(11), 935 – 949
CrossRef - Billones, J. B.; Carrillo, M. C. O.; Organo, V. G.; Sy, J. B.; Clavio, N. A. B.; Macalino S. J. Y.; Emnacen, I. A.; Lee, A. P.; Ko, P. K. L.; Concepcion, G. P.; Drug Des. Devel. Ther. 2017, 11, 563 – 574
CrossRef - Billones, J.B.; Carrillo, M. C. O.; Organo, V.G.; Macalino, S. J.; Sy, J. B.; Emnacen, I. A.; Clavio, N. A.; Concepcion, G. P. Drug Des. Devel. Ther. 2016, 10, 1147 – 1157
CrossRef - Sampaco III, A. B.; Billones, J. B. Orient J. Chem. 2015, 31(4), 1859 – 1865
- Cordova, D. L. M.; Abuel, R. J. D.; Galingana, M. O.; Villanueva, L. A.; Billones, J. B. J. Chem. Pharm. Res. 2015, 7(5), 636 – 642.
- Valle, A. M. F.; Macalalad, E. A.; Abrina, J. L. A.; Cafino, A. J. V.; Pancho, T. M.; Billones, J. B. Phil. J. Health Res. Dev. 2015, 19(1), 1 – 8
- Billones, J. B.; Carrillo, M. C. O.; Organo, V. G.; Macalino, S. J. Y.; Emnacen, I. A.; Sy, J. B. Curr. Enzym. Inhib. 2014, 10(2), 105 – 112
CrossRef - Clavio, N. A. B.; Billones, J. B. J. Chem. Pharm. Res. 2014, 6(10), 727 – 738
- Billones, J. B.; Valle, A. M. F. Orient. J. Chem. 2014, 30(3), 1137 – 1145
CrossRef - Billones. J. B.; Carrillo, M. C. O.; Organo, V. G.; Macalino, S. J. Y.; Emnacen, I. A.; Sy, J. B. A. Orient. J. Chem. 2013, 29(4), 1457 – 1468
CrossRef - Yang, C. T. M.; Billones, J. B. Phil. J. Sci. 2012, 141(2), 187 – 196.
- Uy, V. C. C.; Billones, J. B. Phil. Sci. Lett. 2012, 5(2), 122 – 130.
- Billones, J. B. Orient. J. Chem. 2016, 32(2), 851 – 858
CrossRef - Smith, G.F. In Progress in Medicinal Chemistry, 2009, 1 – 29.
- Banerjee, A.; Dubnau, E.; Quémard, A.; Balasubramanian, V.; Um, K. S.; Wilson, T.; Collins, D.; de Lisle G.; Jacobs, W. R. Jr.; Science 1994, 263(5144), 227 – 230
- Dugan, R. E.; Slakey, L. L.; Porter, J. W.; J. Biol. Chem. 1970, 245(23), 6312 – 6316
- Lu, X.; Lv, M.; Huang, K.; Ding, K.; You, Q. Int. J. Mol. Sci. 2012, 13(6), 6620 – 6638
CrossRef - Rozwarski, D. A.; Grant, G. A.; Barton, D. H.; Jacobs, W. R. Jr.; Sacchettini, J. C.; Science 1998, 279(5347), 98 – 102
CrossRef - Huff, M. W.; Telford, D. E.; Trends Pharm. Sci. 2005, 26(7), 335 – 340
CrossRef - Villagra, A.; Ulloa, N.; Zhang, X.; Yuan, Z.; Sotomayor, E.; Seto, E. J. Biol. Chem. 2007, 282(49), 35457 – 35470
CrossRef - Malumbres, M.; Barbacid, M.; Curr. Opin. Genet. Dev. 2007, 17(1), 60 – 65
CrossRef - Liu, X.; Erikson, R. L.; Proc. Natl. Acad. Sci. U. S. A. 2003, 110(10), 5789 – 5794
CrossRef - Harris, P. S.; Venkataraman, S.; Alimova, I.; Birks, D. K.; Donson, A. M.; Knipstein, J.; Dubuc, A., Taylor, M. D.; Handler, M. H.; Foreman, N. K.; Vibhakar, R. BMC Cancer 2012, 12, 80
CrossRef - Pandey, K. K.; Grandgenett, D. P. Retrovirology, 2008, 2, 11 – 16.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


