High Molecular Weight Poly(Lactic Acid) Synthesized With Apposite Catalytic Combination and Longer Time
Sanjay Kumar Singh, Prashant Anthony and Abhishek Chowdhury
and Abhishek Chowdhury
Department of Chemistry, Sam Higginbottom University of Agriculture, Technology and Sciences, Allahabad-211007, Uttar Pradesh, India.
Corresponding Author E-mail: prashantanthony@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/3404036
Article Received on : 25-05-2018
Article Accepted on : 26-07-2018
Article Published : 20 Aug 2018
Biodegradable polymeric materials derived from renewable resources have attracted tremendous attention in different application. Polylactic acid (PLA) is one of the most useful biodegradable polymers. It is difficult to synthesize high molecular weight polylactic acid by direct polycondensation. The present work describes the synthesis of high molecular weight PLA by varying the duration of reaction and changing the sequence of catalyst addition. Binary catalyst used was SnCl2.2H2O/maleic anhydride and p-toluene sulfonic acid. PLA with molecular weight (6.503X105) was obtained in 50 hours. FTIR, H1 and 13C NMR ascertained the structure of synthesized PLA whereas GPC was used for determining molecular weight.
KEYWORDS:Characterization by FTIR; GPC; H1-NMR; Polylactic acid Synthesis; 13C-NMR
Download this article as:| Copy the following to cite this article: Singh S. K, Anthony P, Chowdhury A. High Molecular Weight Poly(Lactic Acid) Synthesized With Apposite Catalytic Combination and Longer Time. Orient J Chem 2018;34(4). |
| Copy the following to cite this URL: Singh S. K, Anthony P, Chowdhury A. High Molecular Weight Poly(Lactic Acid) Synthesized With Apposite Catalytic Combination and Longer Time. Orient J Chem 2018;34(4). Available from: http://www.orientjchem.org/?p=48691 |
Introduction
In the recent years research attention is shifted on the synthesis and application of biodegradable polymers, which find use in agriculture in the formulation of controlled release fertilizers1 and in increasing the water retention capacity of soil,2 packaging3,4 and biomedical fields.5 The most widely used biodegradable polymers are cellulose, starch, natural rubbers, polylactic acid (PLA), polyhydroxybutyrate (PHA) polycaprolactum (PCL) and poly (butylenes adipate terephthalate) (PBAT).3,6
PLA is the synthetic polymer, which has been a material of interest to most polymer experts. It is a synthetic aliphatic polyester, thermoplastic in nature and produced from renewable resources, as an alternative to petrochemical-derived conventional polymers such as polystyrene (PS), polypropylene (PP), polyethylene (PE) and polyterephthalate (PET). When compared with other biodegradable polymers PLA shows far better properties like mechanical strength, high melting point, high modulus and transparency.3,7,8 However, PLA is seldom used alone without any modification as it has some drawback like slow crystallization, it is brittle in nature, less elongation properties, low heat resistance, limited gas barrier properties and high cost,9,10,11 due to the above said drawbacks the use of PLA is limited only to a few applications. Devanchi and Kaffashi12 has reviewed applications of PLA in diverse medicinal applications like scaffolds, drug delivery systems like microcapsules, microspheres and thin coatings and for surgical sutures and meshes. The applications mainly ascribed to medical field are tissue engineering, artificial organs for temporary or permanent assist, wound management,13 applications in intraocular lens, corneal on lay devices for vision correction the other applications include ureteral stents and stent coatings.14 Polylactic acid nanoparticles have also used for site specific drug delivery,15,16 dermatotherapy17 and many other such medicinal applications.13,18 The physicochemical properties of PLA nanoparticles were affected by molecular weight of PLA.19 It is a great challenge to obtain high molecular weight PLA at a reasonable cost.20,21 High molecular weight PLA has been synthesized by ring opening polymerization of lactide [22, 23] by a multistep process, using catalysts (zinc, lead, tin and aluminium), initiators (n-, sec- and tert-butyl lithium) and solvents (chloroform, toluene, xylene and diphenyl ether) combinations. However, with the use of above reagents for synthesis purification of product is required.23,24 The purification process is difficult and complicated, thereby increasing the cost of PLA. Another process, i.e. direct polycondensation reaction has been also known as melt/solid polycondensation method to produce high molecular weight PLA.20,25-27 This process is completed in two steps, first melt polycondensation and second one solid polycondensation. It was difficult to obtain high molecular weight PLA by direct polycondensation reaction, because the direct polycondensation reaction is reversible reaction, causing equilibrium among free acids, water and finished product thereby making removal of the by-product (i.e. water) difficult. To overcome this problem several approaches have been followed. In one of the study molecular sieve of various pore sizes has been used to remove water from reactions.27,28 In the present work, we will discuss the synthesis of high molecular weight PLA by direct polycondensation at high temperature for a longer time by using a catalyst combination and altering the sequence of catalyst addition and will study the effect of duration of polycondensation reaction on the molecular weight of obtained polymer.
Experimental
Material and Reagents
Lactic acid 90 % extra pure was purchased from Merck, India. Stannous chloride dihydrate (SnCl2. 2H2O) p-Toluene sulfonic acid (TSA), p-xylene, maleic anhydride (MAH), methanol and ethanol were purchased from Central Drug Houses (CDH), India and other reagents used were of analytical grade and were used without further purification.
Method
The synthesis of lactic acid was completed in 500 ml round bottom three necked flask equipped with dean-stark condenser. The polycondensation reaction was carried out by varying time period and changing the sequence of addition of catalyst and keeping a constant temperature under nitrogen atmosphere. The mixture of lactic acid and p-xylene was taken into a round bottom three necked flask. Initially the condensation of lactic acid started without addition of catalyst and reaction was allowed to run for 5 hours at 140 0C temperature. After 5 hours, the reaction temperature was gradually brought down to 100°C then SnCl2.2H2O and maleic anhydride (MAH) corresponding to 0.5% and 0.4% of lactic acid was added in reaction mixture and the reaction was allowed to run for 20 hours at 160°C reaction temperature. After 25 hours, the reaction temperature was further decreased to 100°C and p-toluene sulfonic acid corresponding to 0.5% of lactic acid was added in reaction mixture and reaction temperature was further increased to 160°C and the reaction was allowed to run for different time durations 13, 19 and 25 hours. At the end, the reaction was cooled down to 1000C temperature. The obtained polymeric product was precipitated into methanol and washed 4-5 times with methanol, filtered and dried at 70°C in oven. The synthesized polymeric product was characterized by Gel permeation chromatography (GPC) and spectral analysis such as FTIR, H-1NMR and C-13NMR.
Spectroscopic Analysis
The FTIR spectrum was performed to key out the functional groups and the structure of the synthesized polymer using Perkin Elmer FT-IR spectrum 100 (USA). The absorption peaks in between 4000 cm-1 to 500 cm-1 were studied. H1– NMR spectra of the synthesized polymer samples were scanned on a Bruker Avance II spectrometer working at 400 MHz at 293 K in CDCl3. Chemical shifts (δ) were expressed in ppm. 13C-NMR spectrum of polymer performed using a JEOL AL 300 MHz FTNMR in DMSO solvent. The spectral window was 0 to 200 ppm for 13C-NMR.
Gel Permeation Chromatography
The number-average molecular weight (Mn) and weight average molecular weight (Mw) were determined by Younglin ACME 9000 Gel Permeation Chromatography in DMF at 40 oC with flow rate 0.5 mL/ min on two polystyrene gel columns [PL gel 5µm 10E 4 Ǻ columns (300´ 7.5 mm)] connected in series to a Younglin ACME 9000 Gradient Pump and a Younglin ACME 9000 RI detector. The columns were calibrated against seven poly(methyl methacrylate) (PMMA) standard samples (Polymer Lab, PMMA Calibration Kit, M-M-10).
Results and Discussion
PLA was prepared by polycondensation of lactic acid at 1600C temperature for different time durations and by changing the sequence of catalyst addition. Stannous chloride dihydrate, maleic anhydride and p-toluene sulfonic acid was used as catalysts, SnCl2.2H2O and p-toluene sulfonic acid were used in same ratio corresponding to 0.5% of LA and maleic anhydride was used corresponding to 0.4% of LA. Different samples of PLA were synthesized by direct polycondensation, the molecular weights were determined by GPC method and the obtained results were further confirmed by spectral analysis and structural analysis was determined by FTIR, H1 NMR and 13C NMR.
Fourier Transform Infrared Spectroscopy
The FTIR spectrum of the synthesized polylactic acid (PLA) is shown in figure 1, bands observed at 2998.1 cm-1 and 2948.0 cm-1 can be assigned to symmetric and asymmetric C—H stretching of CH3, and the bands observed at 1455.2 cm-1 can be attributed to methyl/methylene deformation. Band observed at 1758.6 cm-1 can be assigned to the carbonyl group of the ester. The peaks observed at 1189.9 cm-1 can be attributed to C—O stretching in esters.29-31 The absorption band at 870.8 cm-1 can be attributed to the amorphous phase of PLA and the absorption band at 755.7 cm-1 indicate the crystalline phase of PLA.32 The absorption band observed at 3507.5 cm-1 indicate the hydroxyl (–OH) group present in the terminal group of low molecular weight PLA.
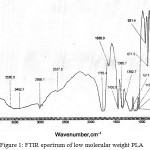 |
Figure 1: FTIR spectrum of low molecular weight PLA.
|
H1 and 13C- Nuclear Magnetic Resonance
The H1-NMR spectrum of the synthesized polylactic acid (PLA) is shown in figure 2, two sets of signals were observed the absorption peaks at 1.527 and 5.14 ppm can be attributed to the proton of CH3 group and CH group present in PLA chain.33 13C-NMR spectrum of PLA is shown in figure 3. The signals at 169.22, 68.75 and 16.52 ppm are representing the carbonyl group of ester bond, CH and CH3 group present in polymeric chain.34
 |
Figure 2: H-1NMR spectrum of PLA synthesized for 50 hours. |
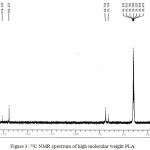 |
Figure 3: 13C NMR spectrum of high molecular weight PLA. |
Gel Permeation Chromatography
Gel permeation chromatography helps in separation of the different components of the mixture based on molecular size. The high molecular weight polymer is eluted first i.e. High molecular weight polymers have the minimum residence time and therefore the time required is less to get eluted, higher peak represents the high molecular weight PLA. Whereas, the low molecular weight polymer spends more time in the pores and therefore slowly eluted, i.e. lower molecular weight polymers needed more time to flow out, and lower peak represents a low molecular weight of PLA.35 According to the given data (Table 1) the molecular weight of PLA increased with the increase of polymerization time duration. The results show when the polymerization was done for 38 hours, (sample A1) 2, 69,800 weight-average molecular weight and 49,200 number average molecular weight of the PLA was obtained, when the polymerization time increased up to 50 hours, (sample A3) molecular weight of PLA obtained increased up-to 6,50,300 weight-average molecular weight and 55,800 number average molecular weight. However, with the increase in reaction time duration polydispersity index was also found to increase.
Table 1: Effect of synthesis time period on the direct polycondensation.
| Sample no. | t(hr) | T (°C) | Mn | Mw | PDI |
| A1 | 38 hr | 160°C | 49,200 | 2, 69,800 | 5.47 |
| A2 | 44 hr | 160°C | 52,700 | 5, 98,300 | 13.08 |
| A3 | 50 hr | 160°C | 55,800 | 6, 50,300 | 14.23 |
The molecular weight data obtained is also supported by FTIR and H1 NMR spectroscopic analysis. With the progress of the polymerization reaction as the reaction time increases the number of hydroxyl group present in polymeric chain decreases, which is reflected by the decrease in intensity of peak at 3507.5 cm-1 attributed to O—H stretching shown in figure 4 whereas the other peaks present in figure 1 and 4 remain almost the same. The decrease in intensity of peak indicate the decrease in amount of hydroxyl group present in polymeric chain which further supports the earlier view of increase of molecular weight of polylactic acid with the increase in reaction time.24,29,36
 |
Figure 4: FTIR spectrum of high molecular weight PLA. |
Figures 5, 6 and 2 show the H1 NMR spectra of the synthesized polymers, on comparing these we find that there are two sets of signals corresponding to CH down field signals were attributed to (5.19, 5.20, 5.21) and due to the CH3 may be assigned to (1.56, 1.59, 1.60) respectively in the samples A1, A2 and A3. The down field shifts in the corresponding samples are indicating higher molecular weights. One δ(CH) at 4.03 which shows a downfield shift as the polymer was increased to 5.19. Another δ (CH3) at 1.23 in monomer that shows a downfield shift because of polymerization increased to 1.46, which is indicative of high molecular weight. Espartero et al.34 has done extensive study on NMR of low and high molecular weight PLA and observed in the case of C13 NMR that there is an upfield shift in the absorption of CO carbon from 176.31 to 169.44 from monomer to polymer. Carbon on CH3 also shows a similar trend whereas CH shows a downfield shift with growth in polymer chain.
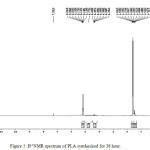 |
Figure 5: H-1NMR spectrum of PLA synthesized for 38 hour. |
 |
Figure 6: H-1NMR spectrum of PLA synthesized for 44 hour. |
Kaitian et al.24 synthesized polylactic acid with two different approaches and found that a high molecular weight polymer is obtained by ring opening polymerization as compared to direct polycondensation; ring-opening polymerization utilizes lactides their production involves several separation and purification steps which results in the high cost of production by this method.27 It has been reported that it is very difficult to obtain high molecular weight PLA by direct polycondensation because of equilibrium between end products and by-products and also for the removal of by-product which is water. In direct polycondensation, it was observed that a linear increase in molecular weight was observed with the increase of temperature upto 200oC. It is observed that with the increase of temperature, the condensation equilibrium constant increases which results in higher molecular weight24 however, with the increase of temperature a higher molecular weight is obtained accompanied by discolouration of the product, which is the result of oxidation.
Fukuzaki et al.21 followed the direct polycondensation route to synthesize polylactic acid in nitrogen atmosphere without the use of catalyst the number average molecular weight Mn increased with the increase of reaction time at 200°C. Initially, at the start of reaction the concentration of reactant was more than the concentration of product. As the reaction proceds, the concentration of reactant decrease and concentration of product increased, which shifts the equilibrium. As a result, formation of PLA decreased when reaction was allowed to run for a longer time without using the catalyst. Therefore, to overcome this problem several researchers have used different organic and inorganic catalyst. Different researchers have tried a wide range of catalyst, the different transition metals and their salts. The various metals employed were tin and its compounds and their combinations viz. powdered tin, stannous oxide, stannous lactate, stannous octoate and stannous chloride, antimony trioxide, titanium oxide, maleic anhydride, succinic anhydride, para toluene sulfonic acid20,21,27,30,37-41 as catalyst. Of the various catalysts tried Yamaoka et al.37 and Kim and Woo39 found stannous chloride to be the most effective and observed that number average molecular weight as high as 40000 can be achieved.
In one of the studies it has been shown that the number average molecular weight of PLA increased with increase in polymerization time up to 20 hours.24 When reaction was allowed to run for longer period the concentration of monomers decreased with increasing molecular weight of PLA. With the decrease in concentration of monomer unit the concentration of terminal group such as hydroxyl and carboxyl group also decreased, leading to the decrease in polarity of the reaction medium which resulted in the change of catalytic activity to favour decomposition.20 This problem can be overcome when binary catalyst such as SnCl2.2H2O/ p-toluene sulfonic acid (TSA), SnCl2.2H2O/ maleic anhydride (MA) and SnCl2.2H2O/ succinic anhydride (SA) used for synthesis of PLA [20, 30, 40]. The proton acids coordinated with vacant coordination sites of catalyst to prevent the activity and reduced the discoloration and depolymerisation.20,40,41,42 When the SnCl2.2H2O/ maleic anhydride (MA) and SnCl2.2H2O/ succinic anhydride (SA) used as binary catalyst near about 150,000 molecular weight of PLA was obtained.30 Therefore when SnCl2.2H2O/ p-toluene sulfonic acid (TSA) used as a binary catalyst the molecular weight increased up to 250,000.20,42 The above results proved that the binary catalyst SnCl2.2H2O/ p-toluene sulfonic acid (TSA) was most effective for the synthesis of higher molecular weight of PLA.
We synthesized high molecular weight of PLA by using SnCl2.2H2O/MAH with TSA proton acid as a catalyst and by altering the sequence of catalyst addition for a longer time period i.e. up to 50 hours. At start of the polycondensation reaction, the rate of formation of polyester increased and that was confirmed by observing the rate of formation of by-product such as water. After some time, the rate of formation of product decreased with the decrease in concentration of reactant. Because of this the catalyst such as SnCl2.2H2O /maleic anhydride was added into reaction mixture. After the addition of the catalyst, the rate of forward reaction increased and suppressed the rate of reverse reaction, due to this the formation of product was increased. But when the condensation reaction was allowed to run for longer time, the concentration of terminal group such as carboxylic acid and hydroxyl group was decreased with the increase in molecular weight of PLA. The terminal group of PLA behaves like a ligand of coordination site of the catalyst. The coordination site of catalyst becomes vacant when the concentration of terminal group decreases because of this the catalytic behaviour gets changed and side reaction such as depolymerisation start, as a result several by-products are formed, causing serious discoloration of product. To avoid this discoloration as soon as it started immediately p-toluene sulfonic acid was added into the mixture. The p-TSA fulfils the vacant site of the catalyst, due to that the catalytic activity was increased. Simultaneously the polymerization reaction and the molecular weight of PLA were also increased. As a result, we were able to obtain approximately 600,000 molecular weight of PLA without discoloration of product in direct polymerization.
Conclusion
High molecular weight polylactic acid synthesized at 160 0C temperature and varying time duration and by changing the sequence of catalyst addition. The synthesized polymers polylactic acid was confirmed by FTIR, H1 NMR and 13C NMR spectral studies. The high molecular weight PLA was achieved by use of multiple catalysts and by adding them at different time. First SnCl2.2H2O and maleic anhydride were added and reaction was allowed to run, after 20 hours of adding the first catalyst p-toluene sulfonic acid was added and reaction was allowed to run for total 13, 19 and 25 hours. The molecular weight of PLA increased with increase in reaction time. GPC data reveals that when the reaction time increased from 38 hours to 50 hours, the weight average molecular weight indicated an increase from 2,69,800 to 6,50,300 and the number average molecular weight also increased from 49,200 to 55,800 whereas the poly dispersity index showed an increasing trend from 5.47 to 14.23. The results obtained were also supported by FTIR and H1 NMR spectral analysis.
Acknowledgment
The authors would like to thank Prof. (Dr) Biswajit Ray, from Department of Chemistry, Banaras Hindu University, Varanasi (India) for carrying out GPC analysis. We are also thankful to SAIF, Panjab University, Chandigarh (India) and Department of Chemistry, Banaras Hindu University, Varanasi (India) for spectral analysis.
References
- Gonzalez, M.E.; Cea, M.; Medina, J.; González, A.; Diez, M.C.; Cartes, P.; Monreal, C.; Navia, R. Sci. Total. Environ. 2015, 505, 446–453
CrossRef - Maghchiche, A.; Haouam, A.; Immirzi, B. J. Taibah. Univ. Sci. 2010, 4, 9-16.
CrossRef - Auras, R.; Harte, B.; Selke, S. Macromol. Biosci. 2004, 4, 835–864
CrossRef - Khosravi-Darani, K.; Bucci, D.Z. Chem. Biochem. Eng. 2015, 29, 275–285.
CrossRef - Duan, Y.; Zhang, Y.; Dong, H.; Zheng, X.; Wang, Y.; Li, H.; Liu, Q.; Zhang, J. Fish. Shellfish. Immunol. 2017, 60, 520-528.
CrossRef - Brigham, C.J.; Sinskey, A.J. Int. J. Biotechnol. Wellness. Ind. 2012, 1, 53-60
- Pillin, I.; Montrelay, N.; Bourmaud, A.; Grohens, Y. Polym. Degrad. Stab. 2008, 93, 321-28.
CrossRef - Carrasco, F.; Pagès, P.; Gamez-Perez, J.; Santana, O.O.; Maspoch, M.L. Polym. Degrad. Stab. 2010, 95, 116-125.
CrossRef - Jaratrotkamjorn, R.; Khaokong, C.; Tanrattanakul, V. J. Appl. Polym. Sci. 2012, 124, 5027–5036.
- Choudhary, P.; Mohanty, S.; Nayak, S.K.; Unnikrishnan, L. J. Appl. Polym. Sci. 2011, 121, 3223–3237.
CrossRef - Ali, F.; Chang, Y.W.; Kang, S.C.; Yoon, J.Y. Polym. Bulletin. 2009, 62, 91–98.
CrossRef - Davachi, S.M.; Kaffashi, B. Polym-Plast. Technol. Eng. 2015, 54, 944-967.
CrossRef - Hamad, K.; Kaseem, M,; Yang, H.W.; Deri, F.; Ko, Y.G. Express. Polym. Lett. 2015, 9, 435–55.
CrossRef - Pawar, R.P.; Tekale, S.U.; Shisodia, S.U.; Totre, J.T.; Domb, J.A. Recent Pat. Regen. Med. 2014, 4,40-51
CrossRef - Leroux, J.C.; Allémann, E.; De Jaeghere, F.; Doelker, E.; Gurny, R. J. Control. Release. 1996, 39, 339–350.
CrossRef - Xing, J.; Zhang, D.; Tan, T. Int. J. Biol. Macromolec. 2007, 40, 153–158.
CrossRef - Rancan, F.; Papakostas, D.; Hadam, S.; Hackbarth, S.; Delair, T.; Primard, C.; Verrier, B.; Sterry, W.; Blume- Peytavi, U.; Vogt, A. Pharm. Res. 2009, 26, 2027– 2036.
CrossRef - Thiyagarajan, D.; Sambandam, B.; Kumar S, S.; Ayyaswamy, A.; Yadav, N. Int. J. Pharm. Pharm. Sci. 2015, 7, 42-49.
- Palacio, J.; Orozco, V.H.; López, B.L. J. Braz. Chem. Soc. 2011, 22, 2304-2311.
- Moon, S.I.; Lee. C.W.; Miyamoto, M.; Kimura,Y. J. Polym. Sci. Part A: Polym. Chem. 2000, 38, 1673–1679.
CrossRef - Fukuzaki, H.; Yoshida, M.; Asano, M.; Kumakura, M. Eur. Polym. J. 1989, 25, 1019-1026.
CrossRef - Lopes, S.M.; Jardini, A.L.; Filho, R.M. Chem. Eng. Trans. 2014, 38, 331-336.
- Khan, F.E.; Van Ommen, J.G.; Feijen, J. Eur. Polym J. 1983, 19, 1081-1088.
CrossRef - Kaitian, X.; Kozluca, A.; Denkbas, E.B.; Piskin, E. Turk. J. Chem. 1996, 20, 43-53.
- Wang, Z.Y.; Zhao, Y.M.; Wang, F.; Wang, J. J. Appl. Polym. Sci. 2006, 99, 244-252.
CrossRef - Ge, Y.P.; Yuan, D.; Luo, Z.L.; Wang, B.B. Express. Polym. Lett. 2014, 8, 50–54.
CrossRef - Pandey, A.K. Int. J. Chem. Res. 2012, 3, 42-51.
- Pandey A.K. Int. J. Adv. Pharm. Bio. Chem. 2013, 2, 68-83.
- Nikolic, L.; Ristic, I.; Adnadjevic, B.; Nikolic, V.; Jovanovic, J.; Stankovic, M. Sensors. 2010, 10, 5063-5073.
CrossRef - Zhang, W.; Wang, Y. Chinese. J. Polym. Sci. 2008, 26, 425−432.
CrossRef - Zhang, J.; Sato, H.; Tsuji, H.; Noda, I.; Ozaki, Y. J. Mol. Struct. 2005, 735-736, 249-257.
CrossRef - Cohn, D.; Younes, H. J. Biomed. Mater. Res. 1988, 22, 993-1009.
CrossRef - Fukuzaki, H.; Yoshida, M.; Asano, M.; Kumakura, M.; Mashimo, T.; Yussa, H.; Imai, K.; Yamanaka, H. Polym. 1990, 31, 2006-2014.
CrossRef - Esoartero, J.L.; Rashkov, I.; Li, S.M.; Manolova, N.; Vert, M. Macromolecules. 1996, 29, 3535-3539.
CrossRef - Yoda, S.; Bratton, D.; Howdl, S.M. Polym. 2004, 45, 7839–7843
CrossRef - Cardoso, J.J.F.; Queirós, Y.G.C.; Machado, K.L.A.; Costa, J.M.; Lucas, E.F. Braz. J. Petrol. Gas. 2013, 7, 057-069.
CrossRef - Yamaoka,T.; Takahashi, Y.; Ohta,T.; Miyamoto, M.; Murakami, A.; Kimura.Y. J. Polym Sci. Part A: Polym. Chem. 1999, 37, 1513–1521.
CrossRef - Shin, G.I.; Kim, J.H.; Kim, S.H.; Kim, Y.H. Korea. Polym. J. 1997, 5, 19-25.
- Kim, K.W.; Woo, S.I. Macromol. Chem. Phys. 2002, 203, 2245-2250.
CrossRef - Khan, G.M.A; Terano, M.; Shamsulalam. M.D. J Polym Mater (2013)5(4) 397-410
- Pivsa-Art, S.; Tong-ngok, T.; Junngam, S.; Wongpajan. R.; Pivsa-Art, W. Energy. Procedia. 2013, 34, 604-609.
CrossRef - Fukushima, T.; Sumihiro, Y.; Koyanagi, K.; Hashimoto, N.; Kimura, Y.; Sakai, T. Int. Polym. Proc. 2000, 7, 380-385.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


