In-Silico Approach Towards Protein Targets Related to Diabets Mellitus-An Overview
N. Abirami and R. Arulmozhi
Department of Chemistry, FSH, SRM University, Kattankulathur-603203, Tamil Nadu.
Corresponding Author E-mail: abirami.n@ktr.srmuniv.ac.in
DOI : http://dx.doi.org/10.13005/ojc/330404
Diabetes mellitus, a chronic disorder which affects people of all ages mainly due to improper food habits, lack of exercise etc. It is difficult to analyze the root cause of this disorder. Laboratory work consumes time and as well requires animal models in order to arrive at good results. Bioinformatics has extended a helping hand through molecular docking for the drug candidate to identify and also to dock with the protein targets related to various diseases. This review paper insights about in silico approach of natural and synthetic compounds which act as both inhibitors or agonists towards protein targets related to diabetes mellitus such as Peroxisome proliferators activated receptor gamma (PPAR-γ), Glycogen synthase kinases 3β (GSK-3β) and Aldose reductase (AR).
KEYWORDS:Diabetes mellitus; animal models; bioinformatics; in silico
Download this article as:| Copy the following to cite this article: Abirami N, Arulmozhi R. In-Silico Approach Towards Protein Targets Related to Diabets Mellitus-An Overview. Orient J Chem 2017;33(4). |
| Copy the following to cite this URL: Abirami N, Arulmozhi R. In-Silico Approach Towards Protein Targets Related to Diabets Mellitus-An Overview. Orient J Chem 2017;33(4). Available from: http://www.orientjchem.org/?p=34729 |
Introduction
Molecular docking is an important method employed for forecasting and investigating the binding capacity between protein targets and drugs. It gives detailed information about the functional groups present in the drug molecules to which the receptor interact and also has created a new coherent approach to drug design1. Diabetes mellitus stands third in life-killing diseases which has attained maximum percentage globally and affects more than 170 million of all ages2. It can be treated in many ways such as insulin injections and hypoglycemic drugs, but still faces long term complications3. All ailments and diseases, involve some biological processes such as, cell-to-cell communication, neural transmission, etc. which is quite complicated to understand the pathway and also to arrive at accurate results. Therefore, controlling diabetes without any ill effects is still a promising experiment to medical field.
In vivo and in vitro studies constitute the experiments for simulating them in the wet lab whereas in silico (computer aided) methods do not need animal models or enzymatic methods. In silico approaches have gained vast popularity recently and have become a vital part of the industrial and academic research that is directed towards drug design and discovery. In medical field, the synergetic combination of computer science and information technology leads to the birth of Bioinformatics. Sequence alignment, gene finding, design of the structure of drug, drugs discovery, protein structure alignment etc4 are the various research domains in bio-informatics.
Drug discovery involves two phases one is discovery of the drug and another is clinical testing both of which demands high costs and time. The final stage involves the approval to market the drug. Before the introduction of in silico studies, the drugs were tested clinically. High throughput screening (HTS) facilitates many chemical, pharmacological tests in speedy manner to identify bio active compounds. In spite of high throughput screening being expensive, it continues to be dynamic and very competitive. A novel high-throughput screening test for proposed anti-diabetic agents interacting with the protein target PPARα was reported5.
This paper gives an overview of in silico approach of various ligands either as inhibitors or agonists on three protein targets related to diabetes mellitus such as Peroxisome proliferators activated receptor gamma (PPAR-γ), Glycogen synthase kinases 3β (GSK-3β) and Aldose reductase (AR). The docking of the ligands with the targets was calculated on the basis of Binding energy (more negative the energy, more the binding).
Peroxisome Proliferator Activated Receptor-Gamma (Pparγ)
Peroxisome proliferator activated receptor-γ is a major regulator in lipid metabolism. There are three types of the PPARγ. They are alpha, beta/delta and gamma. “PPAR alpha is involved in fatty acid uptake in the liver and heart. PPAR beta/delta participates in fatty acid oxidation in muscle. PPAR gamma helps in glucose and lipid uptake, kindles glucose oxidation, decrease free fatty acid level and improve insulin resistance6.”
Peroxisome proliferator-activated receptor gamma (PPARγ) is highly present in adipose tissues that play a significant role on insulin resistance, cell differentiation and energy metabolism. It belongs to the family of nuclear receptor of transcription factors and is an important controller of target genes involved in glucose and lipid homeostasis7.
A drug molecule is considered to be a ligand which binds with protein targets following lock and key mechanism proposed by Emil Fischer, in which the lock represents the protein target and the key is the ligand. The major driving force for binding appears to be hydrophobic interaction which is controlled by hydrogen bonding interactions8.
Recent studies on Peroxisome proliferator-activated receptor-γ reveals that it act as very good receptors for insulin sensitizing drugs.The table 1 revealed the source, type of ligands that docked with various amino acid residues of Peroxisome proliferator-activated receptor-γ, their Binding energies (Kcal/mol) and their Ki in (nM).
Table 1: List of docked ligands to PPARγ receptor
| S.No | Source | Ligands | Interacting Amino acid residues of PPAR γ | BindingEnergy Kcal/mol | Ref.No |
| 1. | Citrus peel | Rosiglitazone | GLN 286, TYR 473, SER 289, HIS 323, HIS 449. ARG 288, CYS 285, LEU 330, MET 329, ILE 326, ALA 292, LEU 469. | -10.46 | 9 |
| Hesperidin | TYR 327, TYR 473, SER 342, GLU 291, HIS 449, ILE 326, ARG 288. CYS 285, LEU 330, SER 289, GLN 286, ILE341, PHE 287. | -9.61 | |||
| Naringin | TYR 473, HIS 323, ARG 288, GLU 291, SER 342, GLU 343. ILE 341, LEU 330, GLN 286, MET 364, CYS 285, GLY 284, SER 289, LEU 270, ILE 326, PHE 287. | -8.68 | |||
| Neohesperidin | LEU 340, GLU 291, GLY 284. GLN 286, SER 289, CYS 285, LEU 330, ILE 341, ARG288, SER 342, GLU 343, VAL339, THR 268, LEU 270, PHE 287. | -8.0 | |||
| Nobiletin | ARG 288, GLU 343. CYS 285, GLY 284, ILE 341, MET 329, LEU 330, SER 342, LEU 228, LEU 333. | -8.22 | 9 | ||
| 2. | Andrographislineata | 5,7,2,3,4-Pentamethoxyflavone | THR266:OG1 THR266:NTHR266:OG1THR266:OG1 | 47.34 | 10 |
| 3. | Andrographisserphyllifolia | 2-Hydroxy-2,4,6-Tri Methoxychalcone | LYS440: OTHR445:NTHR445:OG1 MET452:SD | 40.68 | |
| Dihydroskullcapflavone I | THR266:OG1CYS269:SG THR445: N PHE439:O | 40.2 | |||
| 4. | Andrographislineata | 17,19,20-Trihydroxy-16,15-Olactone | CYS269:SGTHR266:OG1 ASP444:O | 65.66 | |
| 5-Hydroxy-7,8-Dimethoxyflavanone | THR445:N PHE439:OPHE437:OE | 39.04 | |||
| 5-Hydroxy-7,8,2,3,4-Pentamethoxyflavone | CYS269:SGTHR445:N | 32.98 | |||
| 5. | Andrographispaniculata | 5,2-Dihydroxy-7-Methoxyflavanone | LYS440:O MET452:SD MET452:SD | ||
| 5,2-Dihydroxy-7-Methoxyflavanone10 | CYS269:SG | 38.98 | |||
| 6. | Andrographisalata | 5,2-Dihydroxy-7,8-Dimethoxyflavone10 | CYS269:SG | 43.53 | |
| 7. | Andrographislineata | Dimethyl3,3’,4,4’-Tetrahydroxyl- Truxinate10 | SER382:OGLEU419:O | 34.28 | |
| 8. | Roziglitazone [Std] | ASP381:OD2SER382:N | 45.91 | ||
| 9. | Rhizophoraapiculata | Acetamide, N- (4-methylphenyl)- | ILE326 ARG288 | -5.29 | 11 |
| 4-Butyl pyridine, 1-oxide | HIS323 | -5.18 | |||
| Clivorine | ARG288 | -4.27 | |||
| p-Aminodiethylaniline | TYR473 | -4.41 | |||
| 10. | Thiazolidinediones [Std] | TYR473 SER289 HIS449 HIS323 | -4.28 | ||
| 11. | Morindacitrifolia | Rutin | Cys285, Ser289 | -8.5 | 12 |
| Nicotifloroside | Glu291, Ser342 | -8.4 | |||
| Narcissoside | Ser289, Glu343, Met348 | -8.3 | |||
| Quercetin | Ile326 | -8.1 | |||
| Kaempferol | Phe226, Met329, Leu330, Ile326, Arg288, Ala292 | -8.1 | |||
| 12. | Rosiglitazone [Std] | Ser289 | -8.9 | Rosiglitazone [Std] | |
| 13. | Grewiahirsuta | (4Z,12Z)-cyclopentadeca-4, 12-dienone. | Hydrophobic interaction with ILE-268,CYS-432,PHE-439,LEU-694,VAL-342,VAL-265,ILE-345,VAL-349,PHE-346,PHE-313,ILE-310,ALA-272, Polar intact with HIS 435 | Grewiahirsuta | 13 |
Glycogen Synthase Kinase-3β (GSK-3β)
Next promising enzyme to be targeted is Glycogen synthase kinase-3β (GSK-3β). Glycogen synthase kinase-3β which regulates glycogen metabolism and has a control over glucose levels. Glycogen synthase kinase-3β, which is an enthralling enzyme making it an evolving target for dreadful diseases. The GSK-3β inhibitors should be target specific when compared to other enzymes.
“The function of Glycogen synthase kinase-3β is phosphorylating a serine or threonine residue on its target substrate. The phosphate group attached to serine or threonine residues is bonded by a positively charged pocket adjacent to the active site. In turn the active site at residues 181, 200, 97, and 85 binds the terminal phosphate of adenosine tri phosphate and transfers it to the exact location on the substrate14.” The following figure 1 illustrates the above binding.
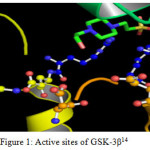 |
Figure 1: Active sites of GSK-3β14 |
Mymoona Akhtar et al15 has carried out the inhibition of Glycogen synthase kinase-3β by involving a novel class of 3-anilino-4-arylmaleimide derivatives in a three-dimensional way. It is achieved by three mechanisms such as Adenosine tri phosphate non-competitive, Adenosine tri phosphate competitive, and metal ion competitive16. “In order to develop models that insights the biological activity of 3-anilino-4-arylmaleimides derivatives, the authors recognized the quantitative structure–activity relationship, comparative molecular similarity index analysis and comparative molecular field analysis models for GSK-3 inhibitors. It revealed strong H-bonding interactions of ligand with amino acid residues Val 135, Asp 133, Arg 141, and Gln185 of GSK-315.” A thorough analysis of both comparative molecular field analysis and comparative molecular similarity index analysis models enabled the authors to give a modification in the structure, which would favor more on inhibition of Glycogen synthase kinase-3β.
Yasser Bustanji et al17 has reported Curcumin as chief curcuminoid constituent present in spice turmeric and it exists as both keto and also as enol form. Mostly enol form is abundantly present. It is used as flavouring and colouring agent. Glycogen synthase kinase exists as two isoforms such as Glycogen synthase kinase-3α and Glycogen synthase kinase-3β, both have similar biochemical functions and substrate affinities18.
The compounds which show inhibition activity against Glycogen synthase kinase-3β have been used as drug candidates for the treatment of many diseases such as type II diabetes, cancer, Alzheimer’s disease, mood disorders, bipolar disorders, stroke, and chronic inflammatory19-23. Currently there are small molecules used as Glycogen synthase kinase-3β inhibitors in clinical studies for the treatment of diabetes mellitus24. Curcumin has conjugated ket-enol system which is hydrogen bonded to VAL-135. The enolic hydroxyl group and the conjugated ketone of curcumin interacts with the amidic carbonyl of VAL-135 (at a separating distance of 3.72A0) and to the NH of the same amino acid residue (at a separating distance of 2.08A0)17.
Karthik Dhananjayan et al25 in their paper studied molecular docking of anthraquinone derivatives on inhibition of Glycogen synthase kinase-3β. The ligands that were used for docking are Aesculin, Aesculetin, Anthragallol, Damnacanthal, Lucidin, Morindone, Nordamnacanthal and Rubiadin. Among the ligands, damnacanthal was found to possess lowest binding energy -7.23kcal/mol, with an estimated inhibition constant (kI) of 4.98uM and nor-damnacanthal showed lowest binding energy of -6.94 kcal/mol with double the value of estimated inhibition constant, kI (8.25 µM). Anthragallol and morindone have similar binding energy values (-6.68kcal/mol) and estimated inhibition constant, kI (12.7 µM) but their conformations at the active site was found to be different. Rubiadin and lucidin has a slight deviation in binding energy at the active site but similar inhibition constant. Aesculetin and Aesculin has binding energy and inhibition constant to be -5.42,-5.99 kcal/mol and 105.78, 40.57 µM respectively. The amino acids involved in binding with the ligands were Asn64, Ser66, Lys85, Val135, Thr138, Lys183, Asp200 and Phe20125. The results revealed that the selective anthraquinone derivatives docks well and thus inhibits the activity of Glycogen synthase kinase-3β25.
In our molecular docking studies13 of inhibitory activity of (4Z,12Z)-cyclopentadeca-4, 12-dienone extracted from the leaves of G.Hirsuta on Glycogen Synthase Kinase 3β revealed that the oxygen atom of the compound forms hydrogen bonding with LYS-85. Strong hydrophobic interaction was observed around the Cyclopentanone ring near the VAL-110, LEU-132, ALA-83, LEU-188, VAL-135, CYS-199 and PHE-67. Polar interactions also occur with amino acids such as Thr 138 and Asn 186. It had binding energy of −6.01 and Gliding energy to be −28.72 respectively13.
Wei Shenet al9 studied phytochemicals such as hesperidin, naringin, neohesperidin, nobiletin present in Citrus flavonoids with the molecular target Glycogen synthase kinase-3β. Hesperidin, naringin, neohesperidin have more OH functional groups in their structures, which might promote the formation of H-bonds between ligands and protein residues. The docking studies of the four ligands with the target showed that citrus flavonoids are potent inhibitors which dock well with the protein targets related to diabetes mellitus and thus impedes its activity. Citrus flavonoids thus play key role in blood glucose regulation, by triggering Peroxisome proliferator-activated receptor-γ, and impeding Glycogen synthase kinase-3β. The more negative binding energy from the table 2 revealed that Hesperidin has good hindering activity towards Glycogen synthase kinase-3β.
Table 2: Docking score of phytochemicals present in citrus fruits with the target GSK-3β
| S.No | List of Phytochemicals | Binding energy |
| 1. | Hesperidin | -9.81 |
| 2. | Naringin | -9.73 |
| 3. | Neohesperidin | -8.3 |
| 4. | Nobiletin | -7.03. |
Aldose Reductase (AR)
Aldo-keto reductase family has one of the chief enzyme Aldose reductase, which is rate limiting enzyme in polyol pathway and reduces glucose to sorbitol by employing Nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) as a cofactor. Sorbitol dehydrogenase is the enzyme responsible for the conversion of Sorbitol into fructose26,27. The polyol pathway signifies a minor route of glucose utilization, which consumes glucose less than 3%. The activity of this pathway is enhanced in the presence of high glucose and takes up to 30% of total glucose consumption28. Evidence for the participation of AR in diabetic neuropathy, retinopathy, nephropathy and cataract arose from several independent studies29. Tissue injury would result, if there is abnormality in this pathway mainly during diabetic condition which in turn leads to accumulation of sorbitol and also oxidative stress30, 31.
Muthuswamy Umamaheswari et al32 has studied the molecular docking of inhibition of flavonoids. The flavonoids that were studied are Farobin-A, GericudraninB, Glaziovianin-A, Rutin, and Xanthotoxin. The standard used in this work was Eparestat, a known aldose reductase inhibitor. The docking results for Eparestat showed binding energy to be -5.59 kcal/mol and all selected flavonoids to be in the range between -7.91 to 5.08 kcal/mol. The active binding sites of the Xanthotoxin were TRP 20, TYR 48, LYS 77, HIS 110, SER 159, ASN 160, GLN 183, GLU 185, TYR 209, SER 210, PRO 211, SER 214, ILE 260 and CYS 298. The binding sites of the Eparestat was found to have amino acid residues such as GLY 18, THR 19, TRP 20, LYS 21, SER 210, PRO 211, LEU 212, SER 214, ILE 260, PRO 261, LYS 262 and SER 263 as active sites. The binding energy, inhibition constant and intermolecular energy of Xanthotoxin and the standard Eparestat were found to be -7.25 kcal/mol and -5.59 kcal/mol, 1.58 µM and 80.08 µM, -7.55 kcal/mol and -5.59 kcal/mol respectively. This implicates that the effective binding sites are present in the selected flavonoid Xanthotoxin and also higher activity when compared with the standard Eparestat32.
Docking analysis was carried out in our previous work on the inhibitory activity of (4Z,12Z)-cyclopentadeca-4, 12-dienone extracted from the leaves of G.Hirsuta with the protein target aldose reductase13. The results revealed that the amino acid residues such as PHE 122, PRO 218, TRP 219, LEU 300, CYS 298, TRP 111, TRP 48, TRP 79, VAL 47, TYR 48 form strong hydrophobic interactions. Polar residues GLN 49and HIS110 present in active site also interact with the compound13. The compound interacts and fits neatly in the hydrophobic active site and induces unique and dramatic conformational changes and has a docking score of −7.61 which implicate that it had good inhibitory activity when compared with standard33.
Akila et al34 using in silico approach aimed to screen the constituents present in Peperomiapellucida, in order to determine the powerful constituent having antidiabetic activity. Docking analysis results clearly indicated that Yohimbine has binding energy of -10.08cal/mol possessing high anti-diabetic activity when compared to the standard, Quercetin (-9.62Kcal/mol).
Arumugam Madeswaran et al35 studied on commercially available flavonoids such as biochanin, butein, esculatin, fisetin and herbacetin on the inhibitory effect of AR. The standard used was Eparestat, a known aldose reductase inhibitor. The results revealed that all the selected flavonoids showed binding energy ranging between -9.33 kcal/mol to -7.23 kcal/mol when compared to that of the standard (-8.73 kcal/mol). Inhibition constant (144.13 µM to 4.98 µM) and intermolecular energy (-11.42 kcal/mol to -7.83 kcal/mol) of the flavonoids also coincide with the binding energy. Butein, biochanin and herbacetin showed excellent binding interactions with aldose reductase enzyme than the standard when compared to other flavanoids. This is due to the difference in the position of the functional groups in the compounds35.
Antony et al36 in their paper investigated on the inhibitory effects of phytochemicals present in dietary spices such as Zingiberofficinale (ginger), Curcuma longa (turmeric) Allium sativum (garlic) and Trigonellafoenumgraecum (fenugreek) on AR. Molecular docking was performed on phytochemicals such as Gingerenones A, B and C, lariciresinol, quercetin and calebin A to study protein-ligand interactions. It is illustrated in the following table 3.
Table 3: Glide Score and interacting residues from XP docking of various phyto compounds and drugs on AR targets.
| S.No | Ligand | Crystal structure of ARTarget | XP Glide score | H-bonding residues |
| 1. | Gingerenone A | 4GCA | -13.02 | Thr113, Gln183 |
| 2. | Gingerenone B | -11.87 | Thr113 | |
| 3. | Quercetin | -11.87 | Leu300, His110 | |
| 4. | Lariciresinol | -11.77 | Gln183 | |
| 5. | Calebin A | -11.76 | Thr113, Gln183 | |
| 6. | Gingerenone C | -11.41 | Thr113, Gln183 | |
| 7. | Ranirestat | -9.7 | Trp111, Tyr48 | |
| 8. | EpARestat | -9.5 | Trp20 | |
| 9. | Sorbinil | -7.4 | Tyr48, His110 | |
| 10. | Gingerenone B | 4LAU | -11.92 | Trp111, Gln183 |
| 11. | Gingerenone A | -11.66 | Thr113, Gln183 | |
| 12. | Quercetin | -11.55 | Thr113 | |
| 13. | Calebin A | -11.13 | Thr113, Asn160, Ala299 | |
| 14. | Lariciresinol | -11.05 | Gln183 | |
| 15. | Gingerenone C | -10.68 | Tyr48, His110, Thr113 | |
| 16. | EpARestat | -9.7 | Trp20 | |
| 17. | Ranirestat | -9.2 | Tyr48, His110 | |
| 18. | Sorbinil | -7.7 | Trp20, Trp111 | |
| 19. | Gingerenone B | 1USO | -11.63 | Tyr48, His110 |
| 20 | Gingerenone A | -11.77 | Thr113, Gln183 | |
| 21. | Quercetin | -11.53 | Thr113 | |
| 22. | Gingerenone C | -11.50 | Thr113, Gln183 | |
| 23. | Lariciresinol | -11.48 | No H bonds | |
| 24. | Calebin A | -11.48 | Thr113, Gln183 | |
| 25. | Ranirestat | -9.7 | Trp111 | |
| 26. | EpARestat | -9.4 | Trp20 | |
| 27. | Sorbinil | -8.7 | Trp111 |
Conclusion
This review paper emphasized about the molecular docking of natural and commercially available compounds with the selected protein targets such as Peroxisome proliferators activated receptor gamma (PPAR-γ), Glycogen synthase kinases 3β (GSK-3β) and Aldose reductase (AR) related to diabetes mellitus. The results gave an insight about possible structures that the ligands could have so that it can dock well with the protein targets. In silico approach would favour the scientists for the discovery of structurally favoured potential compounds for treating the chronic disorder, diabetes mellitus.
References
- Bothara, K.G.; Patil, A.U.; Sexena, A. Importance of docking studies in drug design. Indian .J Pharm. Sci. 1998, 60(6), 333-337.
- Stumvoll, M.; Goldstein, B.J.; Van Haeften, T.W. Type 2 diabetes: Principles of pathogenesis and therapy. Lancet. 2005, 365, 1333-1346.
CrossRef - Satyavati, G.V.; Neeraj, T.; Madhu, S. Indigenous Plant drugs for diabetes mellitus. Dia. Bulletea. 1989, 164Q- 190Q.
- Uma Magheswari, M.; and Sudarsanam, D. A Review on Bio Informatics for Diabetic Mellitus. International Journal of Pharma Sciences and Research. 2012, 3(6), 389-395.
- Hostetler, H.A.; Syler, L.R.; Hall, L.N.; Zhu, G.; Schroeder, F.; Kier, A.B. A Novel High-Throughput Screening Assay for Putative Ant diabetic Agents through PPARα Interactions. J. Biomol. Screen 2008, 13, 855-861.
CrossRef - Limei Wang.; Birgit Waltenberger. Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARγ): a review. Biochemical Pharmacology. 2014, 92(1), 73–89.
CrossRef - Choi, J.H.; Banks, A.S.; Kamenecka, T.M. Antidiabetic actions of a non-agonist PPARγ ligand blocking Cdk5-mediated phosphorylation. Nature. 2011, 477, 477-481.
CrossRef - Hugo, K.Combinatorial and computational approaches in structure-based drug design. Curr. Opin. Drg. Dis. and Develop.1998, 1, 16-27.
- Wei Shen.; Yan-Hua, Lu. Molecular docking of citrus flavonoids with some targets related to diabetes. Bangladesh J Pharmacol. 2013, 8, 156-170.
CrossRef - Sudarshana Deepa.; Suresh Kumar, P. Molecular Docking Studies on selected Phytocompounds from different Andrographis.sp against PPAR γ and C/EBP-α Receptors For Type-2-Diabetes. Asian J. Pharm. Clin. Res. 2013, 6(2), 196-199.
- Gurudeeban Selvaraj.; Satyavani Kaliamurthi.; Ramanathan Thirugnanasambandam. Molecular docking studies on potential PPAR-γ agonist from Rhizophoraapiculata. Bangladesh J. Pharmacol. 2014, 9, 298-302.
CrossRef - Fikry Awaluddin.; Andrianopsyah Mas Jaya Putra.; Supandi. Molecular Docking Studies of Flavonoids of Noni Fruit (Morindacitrifolia L.) to Peroxisome Proliferator-Activated Receptor-Gamma (PPARγ). 3rd International Conference on Computation for Science and Technology (ICCST-3), Atlantis publishing. 2014, 95-99.
- AbiramiNatarajan.; ShobanaSugumar.; SivakumarBitragunta.; Natarajan Balasubramanyan. Molecular Docking Studies of (4Z, 12Z)-cyclopentadeca-4, 12-dien-1-one from Grewiahirsuta with some targets related to Type 2 Diabetes. BMC-Complementary and Alternative medicine. 2015, 15, 73.
CrossRef - Bullock, W.H.; Magnuson, S.R.; Choi. S; Gunn, D.E.; Rudolph, J. Prospects for kinase activity modulators in the treatment of diabetes and diabetic complications. Curr Trop. Med. Chem. 2002, 2, 915–938.
CrossRef - Mymoona Akhtar.; Prasad, V. 3D-QSAR and Molecular Docking Studies on 3-Anilino-4-Arylmaleimide Derivatives as Glycogen Synthase Kinase-β inhibitors. Chem.Biol. Drug Design. 2012, 1-12.
- Van Wauwe, J.; Haefner, B. Glycogen synthase kinase-3 as drug target: from wallflower to center of attention. Drug News Perspect. 2003, 16, 557–565.
CrossRef - Yasser Bustanji.; Mutasem, O. Taha.; Ihab, M. Almasri.; Mohamed, A.S.; Al-Ghussein.; Mohammad, K. Mohammad.; Hatim, S. Alkhatib. Journal of Enzyme Inhibition and Medicinal Chemistry. 2009, 24(3), 771–778.
CrossRef - Stambolic, V.; Woodgett, J.R. Mitogen inactivation of glycogen synthase kinase-3βin intact cells via serine 9 phosphorylation. Biochem. J. 1994, 303, 701–704.
CrossRef - Martinez, A.; Castro, A.; Dorronsoro, I.; Alonso, M. Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neuro degeneration, cancer, and inflammation. Med. Res. Rev. 2002, 22,373–384.
CrossRef - Dorronsoro, I.; Castro, A.; Martinez, A. Inhibitors of glycogen synthase kinase-3: Future therapy for unmet medical needs. Expert Opin.Ther. Patents. 2002, 12, 1527–1536.
CrossRef - Alonso, M.; Martinez, A. GSK-3 inhibitors: Discoveries and developments. Curr. Med. Chem. 2004, 11, 755–763.
CrossRef - Mohammad, M.K.; Al-Masri, I.M.; Taha, M.O.; Al-Ghussein, M.A.; Alkhatib, H.S.; Najjar, S.; Bustanji, Y. Olanzapine inhibits glycogen synthase kinase-3β:An investigation by docking simulation and experimental validation. Eur. J. Pharmacol. 2008, 584,185–191.
CrossRef - Taha, M.O.; Bustanji, Y.; Al-Ghussein, M.A.; Mohammad, M.; Zalloum, H.; Al-Masri, I. M.; Atallah, N. Pharmacophore modeling, quantitative structure–activity relationship analysis, and in silico screening reveal potent glycogen synthase kinase-3β inhibitory activities for Cimetidine, Hydroxy chloroquine, and Gemifloxacin. J. Med. Chem. 2008, 51, 2062–2077.
CrossRef - Dajani, R.; Fraser, E.; Roe, S.M.; Young, N.; Good, V.; Dale, T.C.; Pearl, L.H. Crystal structure of glycogen synthase kinase 3 β: structural basis for phosphate-primed substrate specificity and auto inhibition. Cell. 2001, 105(6), 721–732.
CrossRef - Karthik Dhananjayan.; Sumathy Arunachalam.; Palanisamy Sivanandy. Molecular Docking Studies of Anthraquinone Derivatives on the Crystal Structure of Glycogen Synthase Kinase 3 β. Electronic Journal of Biology. 2014, 10(1), 14-20.
- Ravindranath, T.M.; Mong, P.Y.; Ananthakrishnan. R.; Li, Q.; Quadri, N.; Schmidt, M.A.; Ramasamy, R.; Wang, Q. Novel Role for Aldose Reductase in Mediating Acute Inflammatory Responses in the Lung. J. Immunol. 2009, 183, 8128-8137.
CrossRef - Hwang, Y.C.; Shaw, S.; Kaneko, M. Aldose reductase pathway mediates JAK-STAT signaling: a novel axis in myocardial ischemic injury. J. Fed. Am. Soci. Exp. Biol. 2005, 19, 795-797.
CrossRef - Yadav, U.C.S.; Naura, A.S.; Aguirre, L.A.; Ramana, K.V.; Boldogh, I.; Sur, S.; Boulares, H.A.; Srivastava, S. K. Aldose reductase inhibition suppresses the expression of Th2 cytokines and airway inflammation in ovalbumin-induced asthma in mice. J. Immunol. 2009, 183, 4723-4732.
CrossRef - Guzman, A.; Guerrero, O. R. Inhibition of aldose reductase by herbs extracts and natural substances and their role in prevention of cataracts. Revista Cubana de Plantas Medicinales. 2005, 10, 3-4.
- Dong, Y.; Yang, J.; Ren, X.; Zhang, H.; He, J. New Aldose Reductase Inhibitors N99-596 A and B from Streptomyces. J Antibiotics. 2005, 58, 737-739.
CrossRef - Saraswat, M.; Muthenna, P.; Suryanarayana, P.; Petrash, J.M; Reddy, G. Dietary sources of aldose reductase inhibitors: prospects for alleviating diabetic complications. Asia Pacific J. Clin. Nut. 2008, 17,558-565.
- MuthuswamyUmamaheswari,C.S.;AjiKuppusamyAsokkumar.;ThirumalaisamySivashanmugam.; VaradharajanSubhadradevi.; PuliyathJagannath.; ArumugamMadeswaran. In Silico docking studies of Aldose Reductase Inhibitory activity of selected Flavonoids. International Journal of Drug Development and Research 2012, 4(3), 328-334
- Kinoshita.; Takayoshi. The structure of human recombinant aldose reductase complexed with the potent inhibitor zenarestat. Acta Crystallographica Section D: Biological Crystallography. 2002, 58(4), 622-626.
CrossRef - Akhila, S.; Aleykutty, N.A.; and Manju, P. Docking studies on Peperomiapellucida as antidiabetic drug. International Journal of Pharmacy and Pharmeutical Sciences. 2012, 4(4), 76-77.
- Arumugam Madeswaran.; Muthuswamy Umamaheswari.; Kuppusamy Asokkumar.; Thirumalaisamy Sivashanmugam.; Varadharajan Subhadradevi.; Puliyath Jagannath. In silico docking studies of aldose reductase inhibitory activity of commercially available flavonoids. Bangladesh J Pharmacol. 2012, 7, 266-271.
CrossRef - Antony, P.; Vijayan, R. Identification of Novel Aldose Reductase Inhibitors from Spices: A Molecular Docking and Simulation Study. PLoS One 2015, 10(9), 1-19.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


