Syntheses of Enaminone-Based Heterocyclic Compounds and Study their Biological Activity
Islam H. El Azab1,2,*, Laila M. Break1 and Zainah A. A. El-Zahrani1
1Chemistry Department, Faculty of Science, Taif University, Al-Haweiah, Taif, Saudi Arabia.
2Faculty of Science, Aswan University, Aswan 81528, Egypt.
Corresponding Author E-mail: ihelmy2003@yahoo.com
DOI : http://dx.doi.org/10.13005/ojc/320514
We herein reported the utility of 3-(3-(dimethylamino)acryloyl)-2H-chromen-2-one (2), as a versatile precursor for the synthesis of some new azole systems of possible biological activities. The structure of the newly synthesized compounds has been confirmed by IR, 1H NMR, mass spectral and elemental analysis. Furthermore, some selected compounds were screened in vitro for their anti-microbial activities. The results declared that most of the synthesized compounds have high anti-microbial activity.
KEYWORDS:2H-Chromen-2-one; dimethyformamide-dimethylacetal; enaminone, azole; anti-microbial activity
Download this article as:| Copy the following to cite this article: Azab I. H. E, Break L. M, El-Zahrani Z. A. A. Syntheses of Enaminone-Based Heterocyclic Compounds and Study their Biological Activity. Orient J Chem 2016;32(5). |
| Copy the following to cite this URL: Azab I. H. E, Break L. M, El-Zahrani Z. A. A. Syntheses of Enaminone-Based Heterocyclic Compounds and Study their Biological Activity. Orient J Chem 2016;32(5). Available from: http://www.orientjchem.org/?p=21685 |
Introduction
In our previous work 1,2 , we were interested in the synthesis of enaminones so it is a theme of ongoing interest. Enaminones are versatile precursors that have been utilized extensively as building blocks in organic synthesis 3-12, such compounds are synthetic precursors for the synthesis of a broad diverse of naturally occurring alkaloids 13 and nitrogen-containing heterocycles14.
On other hand, chromene the first heteroaryl in this introduction, exhibited diverse biological activities, and it form the main nucleus of various types of polyphenols and widely found in natural alkaloids, tocopherols, flavonoids, and anthocyanins 15. Natural and synthetic chromenes have potential biological activities such as anti-tumor, anti-vascular16, anti-microbial 17, anti-oxidant 18, TNF-α inhibitor 19, anti-fungal 20, anti-coagulant, anti-spasmolytic, estrogenic 21, anti-viral 22, anti-helminthic, anticancer 23 and anti-HIV 24.
Chromene derivatives are also, play a vital role in the structure of highly effective fluorescent dyes for synthetic fibers, daylight fluorescent pigments and electro photographic and electroluminescent devices 25. Conrauinone A (Fig. 1) was a naturally occurring fused ring chromene, has been isolated from the bark of the tree Millettia conraui and potentially utilized for the treatment of intestinal parasites. Another natural compound was erysenegalensein C (Fig. 1) which has been extracted from the bark of Erythrina senegalensis and found probable use in the treatment of stomach pain, female infertility and gonorrhea 26.
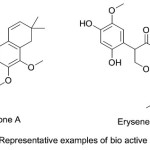 |
Figure 1 |
Finally, as second motif in this presentation, azoles offered unique divergent pharmacological features27. The merge of the heterocyclic nuclei enhances the pharmacological activities than its parent nucleus 28, thus, versatile heterocyclic moieties were appended in the 1,2,4-triazine nucleus were found to be novel therapeutic agents 28. Figure 2 shows some of the reported biologically active 1,2,4-triazine derivatives 29.
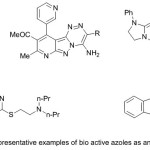 |
Figure 2 |
With these forerunners, we launched a program aiming at merging of multipharmacophores in single architectures30-38. Herein, we describe the synthesis of 2 as polydentate reagent graced with a reactive enaminone tag, which underwent a series of condensations and cyclocondensation manipulations. It was rationale that, the convergence of two pharmacophors in the resulting motifs might evoke eminent pharmacological potencies.
Experimental
Reagents were purchased from Sigma Aldrich and used without further purification. Reaction progress was monitored by TLC on silica gel pre-coated F254 Merck plates. Spots were visualized by ultraviolet irradiation. Melting points were determined on a Gallenkamp electrothermal melting point apparatus and are uncorrected. IR spectra were recorded as potassium bromide discs using Bruker-Vector 22 FTIR Spectrophotometer. The NMR spectra were recorded with a Varian Mercury VXR-300 NMR spectrometer at 300 and 75 MHz for 1H and 13C NMR spectra respectively, using DMSO-d6 as solvents. Chemical shifts were related to that of the solvent. Mass spectra were recorded on a Hewlett Packard MS-5988 spectrometer at 70 eV. Elemental analyses were carried out at the Micro-analytical Center of Cairo University, Giza, Egypt.
General procedure for the reaction of enaminone 2 with primary aromatic amine
A mixture of enaminone 239,40 (1.21 g, 5 mmol), and the appropriate primary aromatic amine (5 mmol) in AcOH acid (20 mL) was refluxed for 5 h. The precipitated product was collected by filtration, washed with MeOH, dried and finally recrystallized from appropriate solvent afforded the corresponding derivatives 3a-d.
3-(3-(Phenylamino)acryloyl)-2H-chromen-2-one (3a)
Yellow crystals, yield: 73%; mp. 245-247 oC (EtOH); IR: 1640 (C=Cstr.), 1695 (C=Ostr.) and 1722 (ring C=Ostr.), 3265 (NHstr.); 1H NMR: 5.72 (d, J = 9.11 Hz , 1H, -CO-CH=), 7.61-8.22 (m, 12H, 11Ar-H and 1H, =CH-N), 10.85 (br, s, 1H, NH, D2O-exchangeable); MS (m/z, %): 291.0 (M+, 15). Anal. Calcd for C18H13NO3 (291.30): C, 74.22; H, 4.50; N, 4.81; O, 16.48%. Found: C, 74.02; H, 4.38; N, 4.69; O, 16.26%.
2-((3-Oxo-3-(2-oxo-2H-chromen-3-yl)prop-1-en-1-yl)amino)benzoic acid (3b)
Yellow crystals, yield: 66%; mp. 263-265 oC (EtOH). IR: 1640 (C=Cstr.), 1695, 1712 (2 C=Ostr.) and 1722 (ring C=Ostr.), 3287 (N-Hstr.); 1H NMR: 5.81 (d, J = 10.01 Hz , 1H, -CO-CH=), 7.61-8.87 (m, 11H, 10Ar-H and =CH-N), 10.98 (br, s, 1H, NH, D2O-exchangeable), 12.06 (br, s, 1H, OH, D2O-exchangeable); MS (m/z, %): 335.0 (M+, 15). Anal. Calcd for C19H13NO5 (335.31): C, 68.06; H, 3.91; N, 4.18; O, 23.86%. Found: C, 67.89; H, 3.71; N, 4.02; O, 23.65%.
3-(3-((2-Nitrophenyl)amino)acryloyl)-2H-chromen-2-one (3c)
Yellow crystals, yield: 85%; mp. 157-159 oC (DMF/ H2O (2:1)); IR: 1358, 1537 (NO2.str.), 1640 (C=Cstr.), 1695 (C=Ostr.) and 1722 (ring C=Ostr.), 3285 (N-Hstr); 1H NMR: 5.81 (d, J = 10.01 Hz, 1H, -CO-CH=), 7.61-8.22 (m, 11H, 10Ar-H and =CH-N), 10.87 (br, s, 1H, NH, D2O-exchangeable); MS (m/z ,%) : 336.0 (M+, 55). Anal. Calcd for C18H12N2O5 (336.30): C, 64.29; H, 3.60; N, 8.33; O, 23.79%. Found: C, 64.12; H, 3.45; N, 8.18; O, 23.58%.
3-(3-((4-Nitrophenyl)amino)acryloyl)-2H-chromen-2-one (3d)
Yellow crystals, yield: 90%; mp. 251-253 oC (DMF/ MeOH (1:3)); IR: 1358, 1537 (NO2.str.), 1640 (C=Cstr.), 1695 (C=Ostr.) and 1726 (ring C=Ostr.), 3285 (N-Hstr.);MS (m/z, %) : 336.0 (M+, 40). Anal. Calcd for C18H12N2O5 (336.30): C, 64.29; H, 3.60; N, 8.33; O, 23.79%. Found: C, 64.15; H, 3.41; N, 8.20; O, 23.61%.
3-(2-Thioxo-2,3-dihydropyrimidin-4-yl)-2H-chromen-2-one (5)
Enaminone 2 (1.21 g, 5 mmol), was added to an ethanolic sodium ethoxide solution, then thiourea (5 mmol) was added. The reaction mixture was refluxed for 8 h. After cooling, the mixture was poured into ice / water mixture. The product that formed was collected by filtration and washed with cold MeOH, then recrystallized from EtOH to give 5 as deep yellow crystals, yield: 65%, mp. 255-257 oC; IR: 1315 (C=Sstr.), 1721 (ring C=Ostr.), 3215 (N-Hstr.); 1H NMR: 5.84 (d, 1H, J = 6.85, pyrim.(C5)-H), 6.65 (d, 1H, J = 6.85, pyrim.(C6)-H),7.20-7.95 (m, 5H, Ar-H), 12.28 (br, s, 1H, NH, D2O-exchangeable); 13C NMR (75 MHz, DMSO-d6): 89.1, 116.2, 118.3, 125.5, 125.7, 127.8, 128.4, 130.7, 153.1, 155.8 (10 C=C), 163.4 (C=NH), 159.6 (C=O), 179.8 (C=S); MS (m/z, %): 256.0 (M+, 45). Anal. Calcd for C13H8N2O2S (256.28): C, 60.93; H, 3.15; N, 10.93; O, 12.49; S, 12.51%. Found: C, 60.73; H, 3.01; N, 10.81; O, 12.29; S, 12.38%.
3-(Benzo[b][1,4]thiazepin-2-yl)-2H-chromen-2-one (10)
A mixture of enaminone 2 (1.21 g, 5 mmol) and o‐amino thiophenol (0.62 mL, 5 mmol) in AcOH acid (25 mL) was refluxed for 8 h. The solvent was partially removed, and the obtained precipitate was filtered off and recrystallized from EtOH to give compound 10 as brown crystals, yield: 63%; mp. 275-277 oC; IR: 1620 (C=Nstr.), 1721 (ring C=Ostr.); 1H NMR: 6.15 (d, 1H, J = 8.45, thiazep.(C3)-H), 6.85 (d, 1H, J = 8.45, thiazep.(C4)-H), 7.25-7.85 (m, 9H, Ar-H); 13C NMR (75 MHz, DMSO-d6): 116.2, 118.2, 125.5, 125.7, 127.8, 128.4, 130.5, 153.1 (8 C=C), 116.3, 117.1, 127.4, 130.3, 138.1, 153.9 (C-Phen.), 99.2, 157.4, 163.9 (C-Thiazep.), 159.5 (C=O); MS (m/z, %): 305.0 (M+, 25). Anal. Calcd for C18H11NO2S (305.35): C, 70.80; H, 3.63; N, 4.59; O, 10.48; S, 10.50%. Found: C, 70.58; H, 3.48; N, 4.37; O, 10.29; S, 10.36%.
3-(3-(Piperidin-1-yl)acryloyl)-2H-chromen-2-one (15)
A mixture of enaminone 2 (1.21 g, 5 mmol) and piperidine (5 mL) in EtOH (30 mL) was refluxed for 5 h, then left to cool. The precipitated product filtered off, washed with EtOH, dried and finally, recrystallized from EtOH /DMF (2:1) yielded compound 15, as pale yellow crystals in 55% yield; mp. 278-280 oC. IR: 1690 (C=Ostr), 1721 (ring C=Ostr.); 1H NMR: 1.54-1.69 (m, 6H, piperid.(C2-C4)-H), 3.21 (t, 4H, piperid.(C1,C5)-H), 5.15 (d, J = 10.12 Hz, 1H, -CO-CH=), 7.20-8.55 (m, 6H, Ar-H), 8.96 (d, J = 10.12 Hz, 1H, =CH-N); MS (m/z; %): 283.0 (M+, 35). Anal. Calcd for C17H17NO3 (283.32): C, 72.07; H, 6.05; N, 4.94; O, 16.94%. Found: C, 71.87; H, 5.79; N, 4.64; O, 16.75%.
3-Oxo-3-(3-(2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)propanenitrile (17)
A mixture of enaminone 2 (1.21 g, 5 mmol) and cyanoacetohydrazide (0.49 g, 5 mmol) in EtOH/AcOH (30 mL) (1:1) was heated under reflux for 4h. During the reflux time, a crystalline solid was separated. The separated solid was filtered off, washed with ethanol to give 17, as deep yellow crystals in 65% yield; mp. 292-294 oC. IR: 1676 (C=Ostr.), 1721 (ring C=Ostr.), 2221 (C≡Nstr.); 1H NMR: 3.61 (s, 2H, CH2), 6.54 (d, 1H, J = 10.12 Hz, Pyr.(C4)-H), 7.20-8.15 (m, 7H, 6Ar-H and Pyr.(C5)-H ); MS (m/z; %): 279.0 (M+, 65). Anal. Calcd for C15H9N3O3 (279.25): C, 64.52; H, 3.25; N, 15.05; O, 17.19%. Found: C, 64.40; H, 3.21; N, 14.89; O, 17.02%.
2-Oxo-4-(2-oxo-2H-chromen-3-yl)-1-phenyl-1,2-dihydropyridine-3-carbonitrile (21)
Sodium ethoxide solution (0.23 g sodium metal in 25 mL absolute EtOH) was added with stirring to a mixture of enaminone 2 (1.21 g, 5 mmol) and 2-cyano-N-phenylacetamide (0.80 g, 5 mmol) in absolute EtOH (25 mL). The reaction mixture was refluxed for 3 h, then poured into cooled H2O (50 mL) and neutralized with diluted HCl acid. The precipitate that formed was filtered off, dried, and recrystallized from the MeOH to afford compound 21, as brown crystals in 65% yield; mp. 158-160 oC. IR: 1650 (amide C=Ostr.), 1723 (ring C=Ostr.); 1H NMR: 5.82 (d, J = 8.12 Hz, 1H, Pyr.(C5)-H), 7.21-7.82 (m, 10H, Ar-H), 8.76 (d, J = 8.12 Hz, 1H, Pyr.(C6)-H); 13C NMR (75 MHz, DMSO-d6): 99.5, 105.5, 116.2, 118.2, 121.2, 125.5, 125.7, 127.9, 128.4, 130.5, 153.1, 171.1 (11 C=C), 128.1, 128.2, 128.9, 139.7 (C-Phen.), 116.1 (C≡N), 158.6, 159.3 (2 C=O); MS (m/z, %): 340.0 (M+, 75). Anal. Calcd for C21H12N2O3 (340.33): C, 74.11; H, 3.55; N, 8.23; O, 14.10%. Found: C, 73.87; H, 3.35; N, 8.01; O, 13.87%.
General procedure of reaction of enaminone 2, with phenols. To a solution of enaminone 2 (1.21 g, 5 mmol) in AcOH acid (15 mL), α-naphthol, β-naphthol (0.72 g, 5 mmol), resorcinol (0.55 g, 5 mmol) or 2-hydroxybenzaldehyde (0.61 g, 5 mmol) was added, then the reaction mixture was refluxed for 12 h. After cooling, the mixture was poured into ice / H2O mixture. The product that formed was collected by filtration, dried and then recrystallized from EtOH to give compounds 25-28.
3-(4H-Benzo[h]chromen-2-yl)-2H-chromen-2-one (25)
Deep brown powder; in 70% yield; mp. 138-140 oC. IR: 1723 (ring C=Ostr.); 1H NMR: 3.36 (d, J = 7.22 Hz, 2H, Pyran.(C4)-H), 5.51 (t, 1H, Pyran.(C3)-H), 6.91-8.12 (m, 11H, Ar-H); MS (m/z, %): 326.0 (M+, 40). Anal. Calcd for C22H14O3 (326.34): C, 80.97; H, 4.32; O, 14.71%. Found: C, 80.75; H, 4.19; O, 14.54%.
2′-Oxo-2′H,4H-[2,3′-Bichromene]-8-carbaldehyde (26)
Reddish brown powder; in 65% yield; mp. 189-191 oC. IR: 1723 (ring C=Ostr.), 1735 (ald., C=Ostr.); 1H NMR: 3.24 (d, J = 7.22 Hz, 2H, Pyran.(C4)-H), 5.53 (t, 1H, Pyran.(C3)-H), 6.91-8.12 (m, 8H, Ar-H), 10.51 (s, 1H, CHO); MS (m/z, %): 304.0 (M+, 15). Anal. Calcd for C19H12O4 (304.30): C, 74.99; H, 3.97; O, 21.03%. Found: C, 74.79; H, 3.71; O, 20.89%.
3-(1H-Benzo[f]chromen-3-yl)-2H-chromen-2-one (27)
As a brown powder; in 75% yield; mp. 158-160 oC. IR: 1723 (ring C=Ostr.); MS (m/z, %): 326.0 (M+, 35). Anal. Calcd for C22H14O3 (326.34): C, 80.97; H, 4.32; O, 14.71%. Found: C, 80.76; H, 4.23; O, 14.49%.
7-Hydroxy-2′H,4H-[2,3′-bichromen]-2′-one (28)
Brown powder; in 85% yield; mp. 139-141 oC. IR: 1723 (ring C=Ostr.), 3451 (br., O-Hstr.); 1H NMR: 3.23 (d, J = 7.22 Hz, 2H, Pyran.(C4)-H), 5.52 (t, 1H, Pyran.(C3)-H), 6.91-8.12 (m, 8H, Ar-H), 10.21 (br, s, 1H, OH, D2O-exchangeable); MS (m/z, %): 292.0 (M+, 25). Anal. Calcd for C19H12O4 (292.29): C, 73.97; H, 4.14; O, 21.90%. Found: C, 73.72; H, 3.86; O, 21.76%.
General Procedure of Reaction of Enaminone 2 With Active methylene compounds.
An equimolar amount of enaminone 2 (1.21 g, 5 mmol), the appropriate amount of acetylacetone, or ethyl acetoacetate (5 mmol) and ammonium acetate (5 mmol) in AcOH (15 mL) was heated under reflux for 6 h. The resulting solid was collected and recrystallized from the proper solvent to give the pyridine derivatives 30a,b.
3-(5-Acetyl-6-methylpyridin-2-yl)-2H-chromen-2-one (30a)
As a yellow crystals in 75% yield; mp. 278-280 oC. IR: 1665 (C=Ostr.), 1712 (ring C=Ostr.); 1H NMR: 2.51 (s, 1H, COCH3), 2.54 (s, 1H, CH3), 7.21-7.75 (m, 5H, Ar-H), 7.96 (d, J = 7.32 Hz, 1H, Pyri.(C3)-H), 8.12 (d, J = 7.32 Hz, 1H, Pyri.(C4)-H); MS (m/z, %): 279.0 (M+, 60). Anal. Calcd for C17H13NO3 (279.29): C, 73.11; H, 4.69; N, 5.02; O, 17.19%. Found: C, 73.01; H, 4.31; N, 4.87; O, 17.02%.
3-(6-Methyl-5-propionylpyridin-2-yl)-2H-chromen-2-one (30b)
As a yellow crystals in 70% yield; mp. 257-259 oC. IR: 1710 (C=Ostr.), 1712 (ring C=Ostr.); 1H NMR: 1.41 (t, 3H, CH2CH3), 2.54 (s, 1H, CH3), 4.12 (q, 2H, CH2CH3), 7.21-7.75 (m, 5H, Ar-H), 7.96 (d, J = 7.32 Hz, 1H, Pyri.(C3)-H), 8.12 (d, J = 7.32 Hz, 1H, Pyri.(C4)-H); MS (m/z, %): 293.0 (M+, 45). Anal. Calcd for C18H15NO3 (293.32): C, 73.71; H, 5.15; N, 4.78; O, 16.36%. Found: C, 73.58; H, 5.01; N, 4.53; O, 16.18%.
3-(5-Hydroxybenzofuran-3-carbonyl)-2H-chromen-2-one (33)
To a stirred solution of the enaminone 2 (1.21 g, 5 mmol) in glacial AcOH (30 mL), 1,4-benzoquinone (0.54 g, 10 mmol) was added and the reaction mixture was stirred overnight at rt. The reaction mixture was evaporated in vacuo, and the solid product obtained was filtered off and finally, recrystallized from EtOH/ DMF (2:1) to afford 33 as deep yellow crystals; yield 78%, mp. 248-250 oC. IR: 1646, 1725 (2 C=Ostr.), 3465 (O-Hstr.). 1H NMR: 6.79-8.22 (m, 8H, Ar-H), 8.91 (s, 1H, furan(C2)-H), 10.05 (br, s, 1H, OH, D2O-exchangable); 13C NMR (75 MHz, DMSO-d6): 116.2, 118.2, 125.5, 127.8, 127.9, 128.4, 139.5, 153.1 (8 C=C), 107.3, 112.7, 112.8, 124.3, 148.8, 151.6 (C-Phen.), 125.9, 160.2 (C-Furan), 159.5, 185.3 (2 C=O); MS (m/z, %): 306.0 (M+, 25). Anal. Calcd for C18H10O5 (306.27): C, 70.59; H, 3.29; O, 26.12%. Found: C, 70.32; H, 3.10; O, 26.01%.
Antimicrobial assessment
Methodology
The antimicrobial activities of the synthesized compounds were tested using the disk-diffusion method41. A plate of 90 mm diameter containing the Muller-Hinton agar for the growth of bacteria and the sabouraud dextrose agar for the growth of fungi were prepared and each plate was separately inoculated with different cultures of the test bacteria and fungi by swabbing aseptically on the whole surface of the agar with cotton wool. A 6 mm diameter filter paper disc was saturated with 200 μg /mL of the tested compounds in DMSO. The discs were air dried and placed aseptically at the center of the plates. The plates were left in refrigerator for 1h before incubation to allow the extract to diffuse into the agar. Ampicillin and gentamicin were used as bacterial standards, while, amphotericin B was used as fungal reference to evaluate the efficacy of the tested compounds, DMSO was used as negative control. After incubation of the plate at the suitable temperature (37 °C for bacteria and 25 °C for fungi), the results were recorded for each tested compound as the average diameter of inhibition zones (IZ) of bacterial or fungal growth around the disks in mm. The minimum inhibitory concentration (MIC) measurement was determined for compounds exhibited significant growth inhibition zones more than 15 mm using two fold serial dilution method42. The MIC (µg/ mL) and IZ diameters values are recorded in Table 1.
Chemistry
In the present study and continuing our previous work 1,2, 30-38 for the synthesis of heterocyclic systems, we report the efficient synthesis of variety of new heterocyclic compounds based on 2H-chromen-2-one, employing the easily obtainable 3-(3-(dimethylamino)acryloyl)-2H-chromen-2-one (2)39,40 as precursor. Thus, the acetyl compound 1 was treated with DMF-DMA, in dry xylene, to afford 3-(3-(dimethylamino)acryloyl)-2H-chromen-2-one (2), (Scheme 1). The reactivity of the enaminone 2 towards some nitrogen nucleophiles was investigated. Thus, the enaminone 2 reacted with some primary aromatic amines in AcOH under refluxing to afford the corresponding acyclic secondary amine derivatives 3a-d, (Scheme 1). The structures of the latter products were confirmed on the basis of their analytical and spectral data. The IR spectra of the products showed C=Cstr., C=Ostr. and NHstr. absorption bands in the regions 1640, 1695- 1722 and 3265-3287 cm-1 respectively. The 1H NMR (DMSO-d6) spectrum of 3b showed two singlet signals corresponding to imino and hydroxyl groups at δ 10.98 and 12.06 respectively, beside a doublet signal due to olefinic proton at δ 5.81 in addition to down field doublet signal due to the other olefinic proton and aromatic protons in the region 7.61-8.87 ppm. Its mass spectrum showed a molecular ion peak at m/z 335.00 (M+, 15%), corresponding to a molecular formula C19H13NO5.
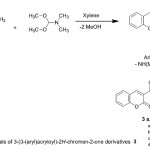 |
Scheme 1 |
The site selectivity in cycloaddition of some nitrogen ampident nucleophiles with enaminones was investigated. Thus, the enaminone 2 treatmented with thiourea afforded a single product (as examined by TLC), for which three possible isomeric cycloadducts 5, 6 and 8, (Scheme 2). The pyrimidine-2-thione isomer 5 was identified as the reaction product based on its elemental and spectral data. Its mass spectrum revealed a molecular ion peak at m/z 256. 00 (M+, 45%), corresponding to a molecular formula C13H8N2O2S. While, its IR spectrum declared absorption band at 1315 cm-1 attributed to (C=Sstr.), besides abroad band at 3215 cm-1 corresponding to (N-Hstr.). Moreover, the observed 1H NMR two doublet signals at 5.84 and 6.65 ppm with coupling constant (J = 6.85 Hz), corresponding to pyrimidine protons, the 1H NMR spectrum of 5 declared a broad singlet at 12.28 ppm attributed to indocyclic imino proton which could only be obtained from isomer 5. Furthermore, its 13C-NMR declared thirteen carbon types, the most important signal is located at 179.8 ppm characteristic for C=S. Compound 5 is assumed to be formed via the addition of one of the NH2 groups of thiourea to the activated exocyclic double bond in the enaminone 2 with subsequent elimination of dimethylamine molecule followed by intramolecular cyclization to annulate the final isolated product 5 via loss H2O molecule.
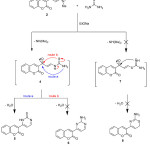 |
Scheme 2 |
Furthermore, the enaminone 2 was cyclocondensed with 2-amino thiophenol in refluxing AcOH to afford 3-(benzo[b][1,4]thiazepin-2-yl)-2H-chromen-2-one (10) via the intermediate 9, which subsequently cyclized through elimination of molecule of water as shown in scheme 3. For this reaction there are three possible isomer products 10, 11 and 13. The structure 11 was easily excluded on the basis of the spectral data of the isolated product. While, both 10 and 13 have the same elemental and spectral data, however, the structure 10 considered as the reaction product so the amino group is the most nucleophilic center than the thiol group. The formation of compound 10 can be explained on the basis of an initial Michael type addition of the exocyclic amino group in 2-amino thiophenol to the activated double bond in enaminone 2 followed by elimination of dimethylamine to afford the non-isolable intermediate 9, which subsequently cyclized through elimination of another molecule of water afforded benzo[b][1,4]thiazepine derivative 10 as shown in scheme 3. The IR spectrum of 10 showed absorption bands at v 1620 (C=Nstr.) and 1721 (C=Ostr.). Its 1H NMR spectrum declared two doublet signals at 6.15 and 6.85 ppm corresponding to azepine protons. The mass spectrum of 10 displayed an intense peak at m/z 305.00 (M+, 25%) corresponding to the expected molecular formula C18H11NO2S.
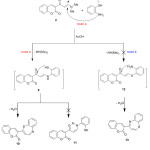 |
Scheme 3 |
In a similar manner, the reactivity of the enaminone 2 towards some secondary amines was studied. Thus, compound 2 reacted with piperidine in refluxing EtOH to afford the corresponding tertiary amine 15, (Scheme 4). The IR spectrum of 15 confirmed the presence of intense absorption bands at 1690 and 1721 cm-1 due to two carbonyl groups. Its mass spectrum showed m/z 283.00 (M+, 35%). Its 1H NMR spectrum showed the presence of multiplet signals at δ 1.54-1.69 (m, 6H, piperid.(C2-C4)-H), 3.21 (t, 4H, piperid.(C1,C5)-H), doublet signal at δ 5.15 and 8.96 due to CO-CH= and =CH-N respectively, and multiplet signal due to an aromatic protons in the region 7.20-8.55 ppm.
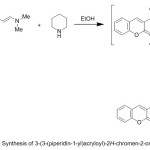 |
Scheme 4 |
The enaminone 2 treated with 2-cyanoacetic acid hydrazide in in refluxing EtOH/ AcOH mixture to afford 3-oxo-3-(3-(2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)propanenitrile 17, (Scheme 5). The structure of the latter product was in agreement with the obtained elemental and spectral data. [cf. experimental part].
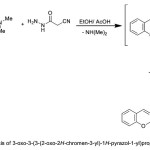 |
Scheme 5 |
The reaction of compound 2 with some active nitriles such as 2-cyano-N-phenylacetamide, in EtONa was studied. Thus, enaminone 2 was reacted with 2-cyano-N-phenylacetamide, to afford 2-oxo-4-(2-oxo-2H-chromen-3-yl)-1-phenyl-1,2-dihydropyridine-3-carbonitrile (21), (Scheme 6). There are two possible isomeric structures for the reaction products 19 and 21. Although, the two theoretically possible structures 19 and 21 have the same elemental analysis and spectral data; the structure 19 ruled out based on the alternate synthesis of the same product 21 via the reaction of 3-acetyl-2H-chromen-2-one with 2-cyano-N-phenylacetamide in refluxing EtOH afforded 2-cyano-3-(2-oxo-2H-chromen-3-yl)-N-phenylbut-2-enamide 22, which underwent cyclocondencation via treating with DMF-DMA afforded the final pyridine derivative 21, (Scheme 6).
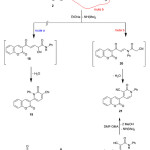 |
Scheme 6 |
The reactivity of the enaminone 2 towards some phenols to attain fused pyran has been investigated. Thus, treatment of enaminone 2 with phenolic group in α–naphthol, 2-hydroxybenzaldehyde, β-naphthol, and resorcinol under AcOH condition afforded the corresponding coumarin derivatives 25-28, (Scheme 7). The structure of these compounds was established on the basis of their elemental analysis and spectral data (IR, 1H NMR and MS). The 1H NMR spectrum of 25 exhibited one doublet signal at δ 3.36 with coupling constant (J = 7.22 Hz), and triplet signal at δ 5.51, corresponding to C4-H and C3-H of pyran ring respectively. The mass spectrum of 25 showed a peak corresponding to the molecular ion at m/z 326.00 (M+, 40%), corresponding to the molecular formula C22H14O3.
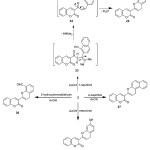 |
Scheme 7 |
Refluxing the enaminone 2 with 2,4-pentanedione or ethyl 3-oxobutanoate in boiling AcOH and AcONH4 afforded pyridine derivatives, 30a,b (Scheme 8). The structures of the products were confirmed by spectral (IR, MS and 1H-NMR) and elemental analyses. The IR spectrum of compound 30a declared the presence of intense absorption bands at 1665 and 1712 cm-1 due to two carbonyl groups. Its, 1H-NMR spectrum revealed two single signals at δ 2.51 (s, 3H, COCH3) and 2.54 (s, 3H, CH3). The mass spectrum of 30a showed a peak corresponding to the molecular ion at m/z 279.00 (M+, 60%), corresponding to the molecular formula C17H13NO3.
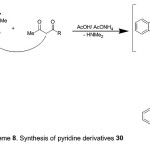 |
Scheme 8 |
Finally, the enaminone 2 reacted with 1,4-benzoquinone in AcOH at rt, yielded 3-(5-hydroxybenzofuran-3-carbonyl)-2H-chromen-2-one (33). The structure of 33 was elucidated based on its elemental analysis and spectral data (IR, 1H NMR and MS). Its IR spectrum showed broad band of the O-Hstr group at 3465 cm-1 and revealed carbonyl absorption bands at 1646 and 1725 cm-1. Its mass spectrum showed a peak corresponding to the molecular ion at m/z 306.00 (M+, 25%), the 1H NMR spectrum of 33 showed characteristic down field singlet signal at δ 8.91 of furan proton and revealed D2O-exchangeable broad singlet at 10.05 ppm due to hydroxyl group [cf. experimental part]. Compound 33 is suggested to be formed via an initial addition of the electron-rich moiety C2 of the enaminone to the activated electron-poor double bond system of the quinone to form the intermediate 31 which readily aromatized and cyclized via dimethylamine elimination into the final isolable product 33, (Scheme 9).
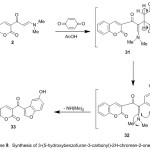 |
Scheme 9 |
Pharmacological Evaluation
Antimicrobial impact
Selected ten newly synthesized compounds 5, 10, 17, 21, 25, 26, 27, 28, 30a and 33 were screened for their in vitro antibacterial activity against two Gram-positive species namely Staphylococcus aureus RCMB 0100010 (SA) and Bacillus subtilis RCMB 010067 (BS) and two Gram-negative species Pseudomonas aeuroginosa RCMB 010043 (PA) and Escherichia coli RCMB 010052 (EC). They were also tested for their in vitro antifungal activity against two fungal species namely Aspergillus fumigatus RCMB 02568 (AF) and Candida albicans RCMB 05036 (CA). Ampicillin was the standard used for the evaluation of antibacterial activity against gram positive bacteria and Gentamicin was used as a standard in assessing the activity of the tested compounds against gram negative bacteria, while Amphotericin B was taken as a reference for the antifungal effect. The inhibitory effects of the synthetic compounds against these organisms are given in Table 1.
The results depicted in Table 1 declared that the most of the screened compounds exhibited variable inhibitory effects on the growth of the tested microorganisms. It is worth noting that, the majority of the tested compounds showed greater potent antibacterial activity against Gram-positive bacteria than Gram-negative. This result is supported by the fact that Gram-negative bacteria have an outer membrane consisting of lipoprotein and lipopolysaccharide, which is selectively permeable and thus control access to the underlying structures. This renders the Gram-negative bacteria generally less susceptible to tested compounds than the Gram-positive bacteria43. It would be also, noticed the compounds belonging to pyrazole, pyridine, pyrimidine, benzo[b][1,4]thiazepine and benzo[b]furan derivatives revealed better antimicrobial potency than the rest of other compounds.
Considering to the structure activity relationship (SAR) of the tested compounds against bacteria, the results declared that compound 17, 21, 30a, 5, 10, 33 and 26 exhibited significant anti-bacterial activity against the tested organisms. Compounds with electron withdrawing groups such as carbonyl, thiocarbonyl or nitrile present in such compounds recorded higher potency than the rest of other compounds. Thus, compounds 17, 21, 30a and 5 were equipotent to Ampicillin in inhibiting the growth of (SA) and (BS) with MIC (4.12 µg/ mL), while, compounds 10, 33 and 26 showed good activity with MIC values (6.25 µg/ mL). On the other hand, compounds 21, 25 and 17 showed moderate potency against Gram-positive bacteria as declared from their MIC values (30.0 µg/ mL). Against Gram-negative bacteria, the data represented in Table 1 revealed that, compounds 17, 21 and 30a were the most potent and were equipotent to Gentamicin in inhibiting the growth of (EC) with MIC values (4.12 µg/mL), and with MIC values (6.25 µg/mL), in inhibiting the growth of (PA). Compounds 5, 10, 33 and 26 exhibited good activity with MIC values (6.25 µg/ mL) in inhibiting the growth of (EC), while, compounds 21, 25 and 28 showed moderate potency against Gram-positive bacteria as declared from their MIC values (30.0 µg/ mL) in inhibiting the growth of (EC). On the other hand, compounds 17, 21, 30a and 5 were the most potent against (PA) with MIC values (6.25 µg/ mL), while the rest of tested compound declared moderate potency as declared from their MIC values (30.0 – 35.0 µg/ mL) in inhibiting the growth of (PA). Also, Table 1 declared that the compounds 17, 21 and 5 showed the highest degree antifungal activity against the tested organisms comparable to Amphotericin B. Compounds 10, 33, 27 and 25 showed moderate antifungal activity against the tested organisms compared with Amphotericin B. While, the compounds 28 and 30a were exhibit the lowest antifungal activity against the tested organisms comparable to Amphotericin B.
Table 1: Antimicrobial evaluation of some synthesized compounds using the disc-diffusion method
| Comp. No.(200 μg/mL) |
MIC values, μg/mLa (Inhibition zone (mm)) |
|||||
|
Gram positive bacteria |
Gram negative e bacteria |
Fungal |
||||
|
SA |
BS |
PA |
EC |
AF |
CA |
|
| 5 |
4.12 (38) |
4.12 (38) |
30.0 (34) |
6.25 (37) |
6.25 (37) |
6.25 (35) |
| 10 |
6.25 (37) |
4.12 (38) |
30.0 (33) |
6.25 (36) |
6.25 (36) |
30.0 (34) |
| 17 |
4.12 (40) |
4.12 (41) |
6.25 (37) |
4.12 (39) |
4.12 (39) |
4.12 (40) |
| 21 |
4.12 (39) |
4.12 (40) |
6.25 (36) |
4.12 (38) |
4.12 (38) |
4.12 (39) |
| 25 |
30.0 (33) |
30.0 (32) |
35.0 (31) |
30.0 (33) |
30.0 (32) |
35.0 (29) |
| 26 |
6.25 (35) |
6.25 (36) |
30.0 (32) |
6.25 (35) |
30.0 (31) |
30.0 (32) |
| 27 |
30.0 (34) |
30.0 (34) |
30.0 (32) |
30.0 (34) |
30.0 (34) |
30.0 (32) |
| 28 |
30.0 (32) |
30.0 (33) |
35.0 (31) |
30.0 (32) |
50.0 (28) |
50.0 (28) |
| 30a |
4.12 (38) |
4.12 (39) |
6.25 (35) |
4.12 (38) |
50.0 (27) |
50.0 (28) |
| 33 |
6.25 (36) |
6.25 (37) |
30.0 (33) |
6.25 (35) |
6.25 (36) |
30.0 (33) |
| Ampicillin |
4.12 (40) |
4.12 (41) |
– |
– |
– |
– |
| Gentamicin |
– |
– |
4.12 (39) |
4.12 (38) |
– |
– |
| Amphotericin B |
– |
– |
– |
– |
4.12 (39) |
4.12 (39) |
| DMSO |
– |
– |
– |
– |
– |
– |
|
a Values are the average of three readings. |
||||||
Conclusion
In this study we report the synthesis and anti-microbial screening of N-heterocycles based on 2H-chromen-2-one. The results indicate that enaminone and N-nucleophiles are useful precursors for the synthesis of different functionalized N-heterocycles. The preliminary in vitro anti-microbial data demonstrated that, the majority of the tested compounds revealed better activity against the Gram-positive rather than the Gram-negative bacteria. The obtained results clearly revealed that compounds derived from enaminone 2 were more potent as anti-microbial agents than enaminone 2
Acknowledgments
The financial support of Taif University, Kingdom of Saudi Arabia and Aswan University, Egypt are gratefully acknowledged.
References
- El-Azab, I.H.; Khaled, M.K. Russ. J. Bioorg. Chem., 2015, 41, 421–436.
CrossRef - El Azab, I.H.; Aly, M.R.E., Heterocycles, 2015, 91, 287-300.
CrossRef - Siddiqui, Z.N.; Ahmed, N.; Farooq, F.; Khan, K. Tetrahedron Lett., 2013, 54, 3599–604.Elassar, A.Z.A.; El-Khair, A.A. Tetrahedron, 2003, 59, 8463-8480.
- Li, M.Y.; Xu, H.W.; Fan, W.; Ye, Q.; Wang, X.; Jiang, B.; Wang, S.L.; Tu, S. J. Tetrahedron, 2014, 70, 1004–1010.
CrossRef - Elassar, A.Z.A.; El-Khair, A.A. Tetrahedron, 2003, 59, 8463-8480.
CrossRef - Stanovnik, B.; Svete, J. Chemical Reviews, 2004, 104, 2433-2480.Gorobets, N.Y.; Yousefi, B.H.; Belaj, F.; Kappe, C.O. Tetrahedron, 2004, 60, 633-8644.
- Yermolayev, S.A.; Gorobets, N.Y.; Lukinova, E.V.; Shishkin, O.V.; Shishkina, S.V.; Desenko, S.M. Tetrahedron, 2008, 64, 4649-4655.
CrossRef - Gorobets, N.Y.; Yousefi, B.H.; Belaj, F.; Kappe, C.O. Tetrahedron, 2004, 60, 633-8644.
CrossRef - Abu-Shana, F.A.; Redhouse, A.D.; Thompson, J.R.; Wakefield, B. J. Synthesis, 1995, 5, 557-560.
CrossRef - Abu-Shanab, F.A.; Aly, F.M.; Wakefield, B. J. Synthesis, 1995, 8, 923-925.
CrossRef - Abu-Shanab, F.A.; Hessen, A.M.; Mousa, S.A.S. J. Heterocycl. Chem., 2007, 44, 4, 787-791.
CrossRef - Abu-Shanab, F.A.; Elnagdi, M.H.; Aly, F.M.; Wakefield, B.J. J. Chem. Soc., Perkin Trans. 1, 1994, 1,1449-1452.
CrossRef - Neto, B.A.D.; Lapis, A.A.M.; Bernd, A.B.; Russowsky, D. Tetrahedron, 2009, 65, 2484–2496.
CrossRef - Hassan, A.A. Inter. J. Org. Chem., 2014, 4, 68-81.
CrossRef - Ren, Q.; Yew Siau, W.; Du, Z.; Zhang, K.; Wang, J. Eur. J. Chem., 2011, 17, 7781–7785.
CrossRef - Gourdeau, H.; Leblond, L.; Hamelin, B.; Desputeau, C.; Dong, K.; Kianicka, I.; Custeau, D.; Bourdeau, C.; Geerts, L.; Cai, S.; Drewe, X.J.; Labrecque, D.; Kasibhatla, S.; Tseng, B. Mol. Cancer Ther., 2004, 3, 1375–1384.
- Chetan, B.S.; Nimesh, M.S.; Manish, P.P.; Ranjan, G.P. J. Serb. Chem. Soc., 2012, 77, 1–17.
CrossRef - Milan, M.; Mirjana, M.; Desanka, B.; Sanja, M.; Neda, N.; Vladimir, M.; Nenad, V.; Slobodan, S.; Slavica, S. Int. J. Mol. Sci., 2011, 12, 2822-2841. Cheng, J.F.; Ishikawa, A.; Ono, Y.; Arrhenius, T.; Nadzan, A. Bioorg. Med. Chem. Lett., 2003, 13, 3647–3650.
- Suresh, T.; Arunima, V.; Atin, K.; Sandeep, G.; Prarthana, V.R.; Ganesh, R.K. Acta Pol. Pharm, 2010, 67, 423-427.
- Nareshkumar, J.; Jiayi, X.u.; Ramesh, M.K..; Fuyong, D.u.; Guo Jian, Z. J. Med. Chem., 2009, 52, 7544–7569.
- Mori, J.; Iwashima, M.; Takeuchi, M.; Saito, H. Chem. Pharm. Bull, 2006, 54, 391-396.
CrossRef - Aliaa, M.K.; Manal, M.K.; Abd El-all, E. K.; AH Elshemy, H. IJPRD, 2012, 4, 310-322.
- Denish, C.K.; Hetal, K.P.; Nilesh, K.G. Asian J. Biochem. Pharma. Res., 2012, 2, 126- 130.
- Khairy, A.M.; Mohsen, M.A.; Yahia, A.M.; Basyouni, W.M.; Samir, Y.A. World J. Chem., 2009, 4, 161-170.
- Jean, W.; Tanee, F.Z.; François, T.; Francine, L.; Michel, K. J. Nat. Prod, 1995, 58, 105–108.
CrossRef - Li, H.; Tatlock , J.; Linton, A.; Gonzalez, J.; Jewell, T.; Patel, L.; Ludlum, S.; Drowns, M.; Rahavendran, S.V.; Skor, H.; Hunter, R.; Shi, S.T.; Herlihy, K.J.; Parge, H.; Hickey, M.;Yu, X.; Chau, F.; Nonomiya, J.; Lewis, C. J. Med. Chem., 2009, 52, 1255–1258.
CrossRef - Abdel-Rahman, R.M.; Makkia, M.S.T.; Alib, T.E.S.; Ibrahim, M.A. Eur. J. Chem., 2010, 1, 236–245.
CrossRef - Kuldip Upadhyay, K.; Manvar, A.; Loddo, R.; La Colla, P.; Virsodiya, V.; Trivedi, J.; Chaniyara, R.; Shah, A. Med. Chem. Res., 2013, 22, 3675–3686.
CrossRef - El-Azab, I.H.; Khaled, M.K. Russ. J. Bioorg. Chem., 2015, 41, 421–436.
CrossRef - El-Azab, I.H.; Abd El Latif, F.M. Lett. Org. Chem., 2015, 12, 187-196.
CrossRef - El-Azab, I.H.; Elkanzy, N.A.A. Synthetic commun., 2014, 44, 2692-2714.
CrossRef - El-Azab, I.H.; El Rady, E.A. Indian J. Chem., 2014, 52(B), 1194-1204.
- El-Azab, I.H. J. Heterocycl. Chem., 2013, 50, 178-188.
CrossRef - El-Azab, I.H.; El Rady, E.A. J. Heterocycl. Chem., 2012, 49, 135-148.
CrossRef - El-Azab, I.H.; Youssef, M.M.; Amin, M.A. Molecules, 2014, 19, 19648-19664.
CrossRef - El-Azab, I.H.; El Rady, E.A. Eur. J. Chem., 2012, 3, 81-86.
CrossRef - El-Azab, I.H.; Elkanzy, N.A.A. J. Heterocycl. Chem., 2016, 00, 000 (Accepted), DOI 10.1002/jhet.2721.
- El-Taweel, F. M. A.; Elnagdi, M. H. J. Heterocycl. Chem., 2001, 38, 981-984.
CrossRef - Al-Zaydi, K.M. Molecules, 2003, 8, 541-555
CrossRef - Bondock, S.; Rabie, R.; Etman, H. A.; Fadda, A. A. Eur. J. Med. Chem., 2008, 43, 2122-2129.
CrossRef - Chung, P. Y.; Chung, L. Y.; Ngeow, Y. F.; Goh, S. H.; Imiyabir, Z. Pharm. Biol., 2004, 42, 292-300.
CrossRef - Chan, E. W. C.; Lim, Y. Y.; Omar, M. Food Chem., 2007, 104, 1586-1593.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


