Effect of CO2 on the Conversion of Isobutane over Iron, Cerium and Molybdenum Mixed Oxides
Daifallah Al-Dhayan, Saad Alqahtani and Ahmed Aouissi
Chemistry Departement, College of Science, King Saud University. Riyadh. Kingdom of Saudi Arabia.
Corresponcing Author E-mail : aouissed@yahoo.fr
DOI : http://dx.doi.org/10.13005/ojc/320535
A series of cerium and iron mixed oxide catalysts were prepared by thermal decomposition of Fe1.5PMo12O40 and Ce1.5PMo12O40 heteropolyanions mixture. The prepared catalysts have been characterized and tested for the conversion of isobutane in the presence of CO2. Characterization by XRD showed that besides Fe2O3 and CeO2, α-MoO3 was the main phase formed after thermal treatment. The effect of the support, the reaction temperature, and the presence of H2O in the reactant mixture was investigated. It has been found that the support enhanced both the conversion and isobutene selectivity. As for the reaction temperature and addition of water, it has been found that increasing the temperature increased both the conversion and isobutene selectivity, whereas the presence of water increased the isobutene selectivity but decreased the conversion.
KEYWORDS:isobutene; isobutene; polyoxometalates; mixed oxides; carbon dioxide
Download this article as:| Copy the following to cite this article: Al-Dhayan D, Alqahtani S, Aouissi A. Effect of CO2 on the Conversion of Isobutane over Iron, Cerium and Molybdenum Mixed Oxides. Orient J Chem 2016;32(5). |
| Copy the following to cite this URL: Al-Dhayan D, Alqahtani S, Aouissi A. Effect of CO2 on the Conversion of Isobutane over Iron, Cerium and Molybdenum Mixed Oxides. Orient J Chem 2016;32(5)..Available from: http://www.orientjchem.org/?p=22542 |
Introduction
Isobutene is one of the most important chemical intermediates used in the production of a variety of products, including the high-octane fuel. The increasing demand for isobutene has drawn considerable attention towards enhancing its production. Currently, it is produced industrially by the dehydrogenation of isobutane (IB) over Cr2O3-Al2O3 catalyst at 627°C 1. Owing to the endothermic nature of this reaction, elevated temperatures are necessary to achieve acceptable levels of conversion. These extreme operating conditions favor coke formation and catalyst deactivation. Furthermore, the formation of carbon oxides is thermodynamically more favorable than the formation of Isobutene. Consequently, the selectivity of Isobutene decreases with the increase of IB conversion when oxygen is used as an oxidizing agent 2, 3. Therefore, much attention has been devoted to develop a new catalyst to make Isobutene production more economical and environmentally friendly. For this reason, several investigations have been carried out on the dehydrogenation of IB in the presence of CO2 4, 6. This reaction is attractive because carbon dioxide is naturally abundant and the resulting process would lead to consumption of a greenhouse gas. However, the Isobutene yield and catalyst stability still major drawbacks for practical applications. Here we report the conversion of IB in the presence of CO2 in gas phase over cerium-iron-molybdenum mixed oxides prepared by thermal decomposition of Ce1.5PMo12O40 and Fe1.5PMo12O40 as heteropolyanion precursors. The effect of the support (Al2O3), cerium molar ratio, reaction temperature, catalyst weight and addition of water on IB conversion was investigated.
Experimental
Preparation of the catalysts
The heteropolyanion precursors, Fe1.5PMo12O40, Ce1.5PMo12O40 used for the preparation of the unsupported and alumina suported mixed oxides catalysts were prepared according to a now well-known method 7. A series of iron-cerium-molybdenum mixed oxides supported on 70% (by weight) of alumina having various Ce/Fe molar ratios have been prepared by incipient-wetness impregnation of Al2O3 with aqueous solutions of Fe1.5PMo12O40 and Ce1.5PMo12O40 in desired volume ratios. The resulting mixture was maintained at about 50°C until dryness evaporation, dried at 100°C for several hours and calcined at 700 °C under an air flow of 6 L/h for 2 h. The prepared catalysts are denoted Ce-Fe-Mo-x where x is the Ce/(Ce+Fe) molar ratio : Ce-Fe-Mo-0; Ce-Fe-Mo-0.33; Ce-Fe-Mo-0.50; Ce-Fe-Mo-0.67.
Characterization of the Catalysts
The Keggin structure of the heteropolyanion precursors and the mixed oxides obtained by thermal treatment of the samples was checked by FT-IR. FT-IR spectra were recorded with an infrared spectrometer GENESIS II-FTIR (4000-400 cm-1) as KBr pellets. The Powder X-ray diffraction (XRD) patterns were recorded on Siemens D5000 diffractometer with Cu-Kα (l = 1.5418Å) radiation using Cu Ka radiation.
Catalytic Measurements
Catalytic tests were carried out under atmospheric pressure using a fixed-bed continuous-flow reactor made of a Pyrex tube. The catalytic charge packed in a stainless reactor, was activated by CO2 flow for 2 h at the reaction temperature. After activation, the reagent mixture CO2 / IB with the desired molar ratio at a flow rate of 50 ml min.-1 was admitted in the reactor. The flow of CO2 and IB gas were controlled by mass flow controller. The products of the reaction were analyzed with a gas phase chromatograph type Agilent 6890 N equipped with a flame ionization detector (FID), a thermal conductivity detector (TCD) and a capillary column (HP-PLOT Q length 30 m ID 0.53 mm).
Results and Discussion
FTIR Characterizations
FTIR spectra of the Keggin-type heteropolyanion precursors, Fe1.5PMo12O40, Ce1.5PMo12O40 used for the synthesis of oxide catalysts are shown in Figure 1. The main characteristic features of the Keggin structure are observed for both catalysts. The four bands characteristic of the kegging structure observed at 1080–1060, 990–960, 900–870, and 810–760 cm-1 are assigned to the asymmetric stretching vibrations of the gas(P–Oa), gas (Mo–Od), gas (Mo–Oc–Mo), and gas (Mo–OC–Mo) bonds, respectively 8, 9. The FTIR spectra of the oxide catalysts obtained by thermal treatment at 700oC are shown in Figure 2. It can be seen that after thermal treatment the typical bonds of the Keggin structure disappeared and replaced by characteristic bonds of Mo-oxo species closely resembling the orthorhombic molybdenum trioxide, referred to as a-MoO3.
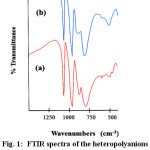 |
Figure 1: FTIR spectra of the heteropolyanions precursors. (a) Fe1.5PMo12O40 (b) Ce1.5PMo12O40 |
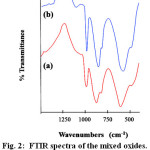 |
Figure 2: FTIR spectra of the mixed oxides. (a) Fe-Mo-oxides (b) Ce-Mo-oxides
|
XRD Measurements
The XRD patterns of the obtained iron molybdenium mixed oxides (denoted as Fe-Mo-oxides) and cerium molybdenium mixed oxides (denoted as Ce-Mo-oxides) are shown in Figure 3 and presented in Table 1. Table 1 presents the d-spacings that correspond to the most significant peaks in the patterns of the mixed oxides Fe-Mo and Ce-Mo and the orthorhombic α-MoO3 oxide 10. These results showed that besides the iron and cerium oxides resulting from the corresponding cation (Fe and Ce, in the precursor heteropolyanions, the mixed oxides contain α-MoO3 as main phase. The diffraction peaks observed at 3.006 Å, 2.702 Å, and 1.632 Å which are ascribed to (111), (200), (220) crystal planes are characteristic of the cubic CeO2 (JCPDS Card No. 43-1002) 11. The reflections observed with d spacing at 3.44 Å, 3.25 Å, 3.07 Å, and 2.69 Å (JCPDS No. 30-0304) are characteristics of Ce2Mo4O15. The indexed XRD patterns for the iron oxides showed that the Mo-Fe catalyst contains peaks of rhombohedral α-Fe2O3 at d-values 2.67, 1.44, (matched with PDF No-01-079-007) and the orthorhombic ferric molybdate (Fe-MoO3) 12. For both iron and cerium mixed oxides, other phases could be present as traces. It is worth noting that for several oxides the coexistence of various phases besides the main phase has been observed 13, 14.
Table 1: Interplanar Spacings dhkl (A°) and Relative Intensities of Orthorhombic α-MoO3 (Ref. 15): Comparison with the XRD Patterns of Fe-Mo-oxides and Ce-Mo-oxides.
____________________________
d-spacings (A°)
___________________________________
|
α-MoO3 |
Mo-Fe |
Mo-Ce |
|
6.942 |
7.070 |
6.901 |
|
3.810 |
4.585 |
4.114 |
|
3.462 |
3.835 |
3.807 |
|
3.260 |
3.501 |
3.464 |
|
2.653 |
3.280 |
3.260 |
|
2.308 |
3.018 |
3.006 |
|
2.670 |
2.881 |
|
|
2.555 |
2.702 |
|
|
1.452 |
2.143 |
|
|
1.437 |
1.988 |
|
|
1.579 |
||
|
1.482 |
||
|
1.449 |
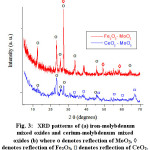 |
Figure 3: XRD patterns of (a) iron-molybdenum mixed oxides and cerium-molybdenum mixed oxides (b) where o denotes reflection of MoO3, à denotes reflection of Fe2O3, denotes reflection of CeO2.
|
Catalytic Activity
The mixed oxide Ce-Fe-Mo-0.67 prepared by chemical and mechanical mixing have been tested in the conversion of IB. The variation of the IB conversion as a function of time on stream is shown in Figure 4. It can be seen from these results that both catalysts have the same catalytic properties. Indeed, we notice that both catalysts led to the same values of conversion and the same stability during the reaction time. It is also noted that they led to the same selectivities of the products (Figure 5). It is worth noting that for each of the catalysts, the selectivity in isobutene decreases with time in favor of the selectivity of cracking products.
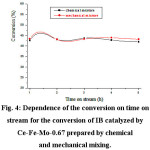 |
Figure 4: Dependence of the conversion on time on stream for the conversion of IB catalyzed by Ce-Fe-Mo-0.67 prepared by chemical and mechanical mixing. |
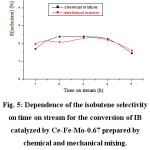 |
Figure 5: Dependence of the isobutene selectivity on time on stream for the conversion of IB catalyzed by Ce-Fe-Mo-0.67 prepared by chemical and mechanical mixing. |
Effect of Support
Figure 6 showed the IB conversion and product selectivites obtained over unsupported Fe-Ce-Mo catalyst and Alumina supported Fe-Ce-Mo. The obtained conversion and isobutene selectivity were 5.54% and 26.9% respectively whereas those obtained over the alumina supported catalyst are (5.79%) and selectivity (28.8%) respectively. These results indicated that the alumina support has slightly improved the catalyst performance. This result indicated that the oxidative dehydrogenation leading to the isobutene formation required the presence of Lewis acid sites.
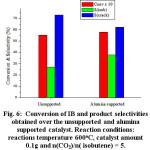 |
Figure 6: Conversion of IB and product selectivities obtained over the unsupported and alumina supported catalyst. Reaction conditions: reactions temperature 600oC, catalyst amount 0.1g and n(CO2)/n( isobutene) = 5. |
Effect of Ce/Fe Ratio
The series of alumina supported Fe-Ce-Mo-x catalysts were tested for the conversion of IB. The dependence of the conversion and the product selectivities on Cerium molar ratio at the steady state are reported in Figure 7. The results showed that the highest conversion of IB (5.8%) and the highest selectivity of isobutene (28.8%) were obtained over Fe-Ce-Mo-0.67 catalyst. In contrast the lowest conversion (4.7%) and the highest selectivity of isobutene (21.2%) were obtained over Fe-Ce-Mo-0.50 catalyst. Since the Fe-Ce-Mo-0.67 catalyst was the best catalyst, it was selected for further investigations such as the effect of the catalyst amount, reaction temperature and addition of water on the IB conversion.
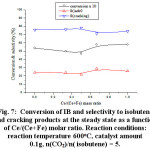 |
Figure 7: Conversion of IB and selectivity to isobutene and cracking products at the steady state as a function of Ce/(Ce+Fe) molar ratio. Reaction conditions: reaction temperature 600oC, catalyst amount 0.1g, n(CO2)/n( isobutene) = 5.
|
Effect of the Catalyst Amount
The variation of the conversion and selectivities as a function of the mass of the catalyst is depicted in Figure 8. It can be seen from the figure that both the conversion of IB and the selectivity of isobutene increased with the increase of the mass of the catalyst. Indeed the conversion increased from 5.8% to 7.5% and the selectivity of isobutene increased from 28.8% to 39.9% when the mass of the catalyst was increased from 0.1g to 0.9 g. In contrast the selectivity of cracking products decreased with the increase of the mass of the catalyst. This result can be explained by the fact that at higher catalyst mass the residence time is long which allow to the dehydrogenation reaction to occur whereas at lower catalyst mass, the residence time is short which decrease the probability for the dehydrogenation reaction to occur and thermal cracking reactions occur.
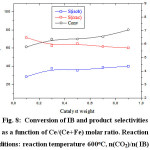 |
Figure 8: Conversion of IB and product selectivities as a function of Ce/(Ce+Fe) molar ratio. Reaction conditions: reaction temperature 600oC, n(CO2)/n( IB) = 5. |
Effect of Reaction Temperature
The Figure 9 shows the dependence of the IB conversion and the product selectivities on the reaction temperature. As expected the conversion increased with increasing the temperature. As for the selectivity, the results showed that the isobutene selectivity increased with the temperature whereas that of cracking products decreased. The isobutene selectivity increased from 26.0% to 39.7% when the temperature increased from (575oC) to (600oC). Beyond 600oC, the selectivity of both isobutene and cracking products remained almost stable whereas the conversion still increasing. This result can be explained by the fact that at higher temperatures, CO2 removes the carbon deposited (responsible of the catalyst deactivation) from the catalyst surface. In fact, it has been reported that the regeneration of deactivated Ni/La2O3 and Ni/Y2O3 catalysts was due to the partial removal of carbon deposits through CO formation (reverse Boudouard reaction) 15-19. The increase of the selectivity of isobutene form 575 oC to 600oC is in agreement with those reported in the literature [20] where it has been mentioned that the temperature for the carbon dioxide to be activated over VMgOx catalyst was around 527–553 oC. CO2 enhanced the yield of IB dehydrogenation significantly. In the opinion of the authors, CO2 as a weak oxidant can eliminate hydrogen produced during the dehydrogenation through the reverse water gas shift (RWGS) reaction 21-23:
C4H10 → C4H8 + H2
CO2 + H2 → CO + H 2O
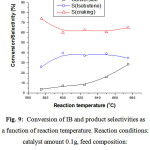 |
Figure 9: Conversion of IB and product selectivities as a function of reaction temperature. Reaction conditions: catalyst amount 0.1g, feed composition: n(CO2)/n(isobutane) = 5 |
Effect of Addition of Water
The effect of water introduction as reactant besides CO2 on IB conversion was also investigated. The reactions were carried out at 600oC over Fe-Ce-Mo-0.67 catalyst with a reactant mixture n(CO2)/n(IB) = 3. The dependence of the conversion and the product selectivities on water proportion is presented in Figure 10. The results showed that the addition of water decreased both the conversion and cracking products but increased the isobutene selectivity. Indeed, when the (nH2O/n CO2) ratio increased from 0.1 to 0.61, the conversion decreased from 5.66% to 21.3% whereas the isobutene selectivity increased form 16.3% to 33.8% to. This result indicated that the presence of water in the reactant mixture favors the oxidative dehydrogenation of IB leading to the formation of isobutene, whereas CO2 favors the formation of the cracking products.
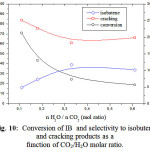 |
Figure 10: Conversion of IB and selectivity to isobutene and cracking products as a function of CO2/H2O molar ratio.
|
Conclusion
Iron, cerium and molybdenum mixed oxides catalysts prepared by thermal treatment of Keggin-type heteropolymolybdates as precursors were characterized and tested for the conversion of IB in the presence of CO2.
Characterization of the catalysts by XRD showed that besides iron and cerium oxides the catalysts contain α-MoO3 as main phase.
The effects of various parameters such as the support (Al2O3), the catalyst weight, the reaction temperature and the presence of water in reactant mixture were investigated. It has been found that Al2O3 improved both the conversion and isobutene selectivity which indicated that the oxidative dehydrogenation leading to the isobutene formation required the presence of Lewis acid sites.
IB reaction over different amounts of showed that both the conversion of IB and selectivity of isobutene increased with increasing the mass of the catalyst.
The dependence of the IB conversion on the temperature showed that beyond 600oC, CO2 enhanced the yield of IB dehydrogenation significantly.
The presence of water in the reactant mixture favors the oxidative dehydrogenation of IB leading to the formation of isobutene, whereas CO2 favors the formation of the cracking products.
Acknowledgment
The Authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding the work through the research group project No RGP-VPP-025
References
- Matsuda, T.; Koike, I.; Kubo, N.; Kikuchi, E. Appl Catal A. 1993, 96, 3-13
- Al-zahrani, S. M.; Elbashir, N O.; Abasaeed, A. E.; Abdulwahed M. J Mol Catal A. 2004, 218, 179-186
- Li, L.; Yan, Z F. Progr Chem. 2005, 17, 651-659
- Dias.; C. R.; Zavoianu, R.; Portela, M. F. Catal Commun. 2002, 3, 85-90
- Bi, Y. L.; Zhen, K. J.; Valenzuela, R. X.; Jia, M. J.; Corberán V. C. Catal Today. 2000, 61, 369-375
- Ogonowski, J.; Skrzńska, E. Catal Lett. 2006, 111, 79-85
- Aouissi, A.; Apblett, A. W.; AL-Othman, Z. A.; Al-Amro, A. Transition Met Chem. 2010, 35, 927-931.
- Rocchiccioli-Deltcheff, C.; Fournier, M.; Franck, R.; Thouvenot, R. Inorg. Chem. 1983, 22, 207-216
- Rocchiccioli-Deltcheff, C.; Fournier, M. J. Chem. Soc. Faraday Trans. 1991,87, 3913-3920
- Andersson, G.; Magnéli, A. Acta Chem. Scand. 1950, 4,793-797
- Huang, S.; Liu, S.; Zhu, Q.; Zhu, X.; Xin,W.; Liu, H.; Feng, Z.; Li, C.; Xie, S.; Wang, Q.; Xu, L. Appl Catal A. 2007, 323, 94-103
- Massarotti, V.; Flor, G.; Marini, A. J Appl Cryst. 1981, 14, p. 64-65
- Carriazo, J. G.; Molina, R.; Moreno, S. Appl Catal A. 2008, 334, 168-172
- Rashad, M. M. Mater Sci Eng B. 2006, 127, 123-129
- Bednarczuk, L.; Ramírez de la Piscina, P.; Homs, N. Int. J. Hydrogen Energy. 2015, 40, 5256-5263
- Jiang, N.; Burri, A.; Park, S. E. Chinese Journal of Catalysis. 2016, 37, 3–15
- Irun, O.; Sadosche, S. A.; Lasobras, J.; Soler, J.; Frances, E.; Herguido, J.; Menendez, M. Catal. Today, 2013, 203, 53-59.
- Hong, D.Y.; Vislovskiy, V. P.; Hwang, Y. K.; Jhung, S. H.; Chang, J. S. Catal. Today, 2008, 131, 140-145.
- Burri, A.; Jiang, N.; Yahyaoui, K.; Park, S. E. Appl. Catal. A. 2015, 495,192-199.
- Ogonowski, J.; Skrzyńska, E. Catal Commun. 2009, 11, 132-136
- Ding, J. F.; Qin Z. F.; Li X. K.; Wang G. F.; Wang J. G.; J Mol Catal A. 2010, 315,221-225
- Shimada, H.; Akazawa, T.; Ikenaga, N.; Suzuki, T. Appl Catal A. 1998, 168, 243-250
- Sun, A. L.; Qin, Z. F.; Chen, S. W.; Wang, J. G. J Mol Catal A. 2004, 210, 189-195
- Ping, H.; Kou, Z.; Xu, G.; Wu, S. J. Environmental Chemical Engineering. 2016, 4, 3253–3259

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


