A Newly Improved Modified Method Development and Validation of Bromofenac Sodium Sesquihydrate in Bulk Drug Manufacturing
Sunil Kumar Yelamanchi V 1, Useni Reddy Mallu2 , I. V Kasi Viswanath1*, D. Balasubramanyam3 and G. Narshima Murthy4
1Department of Chemistry, K.L.University, Green Fields, Vaddeswaram, Guntur, A.P, India. 2Celltrionpharm,inc .Seoul, South Korea . 3M/S Dr.Reddys laboratories limited, Head of quality Control, A.P, India. 4M/S Hetero-labs. limited, Andhra Pradesh., Head of quality Control, A.P, India. Corresponding Author E-mail: Viswanath_fed@kluniversity.in
DOI : http://dx.doi.org/10.13005/ojc/320502
The main objective of this study was to develop a simple, efficient, specific, precise and accurate newly improved modified Reverse Phase High Performance Liquid Chromatographic Purity (or) Related substance method for bromofenac sodium sesquihydrate active pharmaceuticals ingredient dosage form. Validation of analytical method is the confirmation by examination and the provision of objective evidence that the particular requirements for a specific intended use are fulfilled as per ICH, USP, BP or any other suitable regulatory guidelines. The Reverse Phase High Performance Liquid Chromatographic Gradient method was developed by utilizing Waters Symmetry C8, 150x4.6mm, 3.5µm on Waters 2487 series Liquid Chromatograph. The retention time of bromofenac sodium sesquihydrate was found to be 5.973minutes. Considering all the results of validation parameters simplicity of the method and the cost effectiveness of the overall procedure, it is possible to conclude that the developed method can be suitable for the regular quality control determination of bromofenac sodium sesquihydrate in bulk as well as pharmaceutical dosage form.The developed Reverse Phase High Performance Liquid Chromatographic Purity (or) Related substance method for bromofenac sodium sesquihydrate active pharmaceuticals ingredient method was validated with respect to System Suitability , linearity, , precision ,Range, Ruggedness, Test Solution and Mobile phase stability ,Robustness..
KEYWORDS:Bromofenac sodium sesquihydrate; Reverse Phase High Performance Liquid Chromatographic Purity (RPHPLC); Determination; Validation; Pharmaceutical dosage form
Download this article as:| Copy the following to cite this article: Yelamanchi V S. K. Y, Mallu U. R, Viswanath I. V. K, Balasubramanyam D, MurthyG. N. A Newly Improved Modified Method Development and Validation of Bromofenac Sodium Sesquihydrate in Bulk Drug Manufacturing. Orient J Chem 2016;32(5). |
| Copy the following to cite this URL: Medvedev V, Vaniyev M, Sakibayeva S, Kovaleva A. Y. Operating Characteristics of Diene-Urethane Elastomers on the base of NISSOG 3000 Oligodienediol. Orient J Chem 2016;32(5). Available from: http://www.orientjchem.org/?p=21463 |
Introduction
Bromofenac sodium sesquihydrate (which is the sodium salt of 2-amino-3-(4-bromobenzoyl)phenyl acetic acid, also sometimes referred to as sodium 2-amino-3-(4-bromobenzoyl)phenyl acetate sesquihydrate, having the empirical formula C15H11BrNNaO3.1.5H2O) in Figure-1 is a non-steroidal anti-inflammatory drug (NSAID) with analgesic properties. It was initially marketed as an oral suspension under the trade name DURACT but was withdrawn from the United States market in 1998 due to drug-induced hepatotoxicity leading to acute liver failure. Currently, bromfenac sodium is sold as an ophthalmic solution under the brand name XIBROM. The Food and Drug Administration approved this product in 2005 for use in ophthalmic surgery including postoperative inflammation, reduction of pain after cataract and refractive surgery and management of macular edema after cataract surgery. XIBROM ophthalmic solution contains 1.035 mg bromfenac sodium, equivalent to 0.9 mg bromfenac free acid per mL of solution, giving a 0.09% sterile topical ophthalmic formulation with a pH of 8.3. Anti-inflammatory agents that are non-steroidal in nature. In addition to anti-inflammatory actions, they have analgesic, antipyretic, and platelet-inhibitory actions.They act by blocking the synthesis of prostaglandins by inhibiting cyclooxygenase, which converts arachidonic acid to cyclic endoperoxides, precursors of prostaglandins. Inhibition of prostaglandin synthesis accounts for their analgesic, antipyretic, and platelet-inhibitory actions; other mechanisms may contribute to their anti-inflammatory effects. See a list of PubChem compounds matching this category.1-11
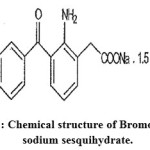 |
Figure 1: Chemical structure of Bromofenac sodium sesquihydrate.
|
Literature review reveals that few spectrophotometric and LC, LCMS methods have been reported for analysis of Bromofenac sodium sesquihydrate. LC, LCMS simultaneous methods for pharmaceutical dosage and one for injections were reported in the literature review; existing methods were Long cyclic run time chromatographic process method. More over reported methods were not much cost-effective in terms of solvent consumption and total run time of the analysis, so we decided to perform rapid, selective and precise newly improved modified method development and validation of Reverse Phase High Performance Liquid Chromatographic Gradient method for determination of Related impurities present in the Bromofenac sodium sesquihydrate in solid dosage form.12-13.
Materials and Methods
Chemicals and Reagents
Reference standard of Bromofenac sodium sesquihydrate ,Related Impurities are 7-(4-bromobenzoyl)-1,3-dihydro-indol-2-one(Bro-4), (4-Bromo phenyl)-(1H-indol-7-yl) methanone (Bro-2),(3-Bromo-1H-indol-7-yl)-(4-bromo-phenyl)-methanone(Bro-3) & (4-Bromo-Phenyl)-(2,3-dibromo-1H-indol-7-yl)-methanone (Dibromo Impurity) and samples was obtained from well reputed research laboratories and characterized by use of LCMS, NMR and IR . All the chemicals were analytical grade from Rankem Ltd., Mumbai, India, while Methanol, Acetonitrile (HPLC grade), Potassium dihydrogen phosphate and Ortho phosphoric acid (HPLC grade) procured from Merck Pharmaceuticals Private Ltd., Mumbai, India and purchased from Merck Specialties Private Ltd., Mumbai, India. The Liquid Chromatography system was equipped with quaternary gradient pumps with auto sampler and column oven , auto injector connected to a variable wave length programmable ultra Violet visible detector all were controlled by Empower software and Manufactured by Waters Alliance system & 2489 UV detector .
Optimization of mobile phase
Optimization of mobile phase was performed based trial and error method. In this different mobile phase trial was taken like in methanol :water, ACN: water and methanol: ACN: water in different ratio without pH but there are different problem were observed like high tailing factor value and not optimized theoretical plate. When the mixture of Phosphate buffer (pH adjusted to 3.2 using dill. Ortho phosphoric acid) and Acetonitrile , Methanol in ratio of 80:10:10 v/v was selected as mobile phase-A and Acetonitrile ,Water in the ratio of 90:10 as mobile phase-B in gradient form Bromofenac sodium sesquihydrate full fill all the criteria of system suitability. The Gradient mobile phase –A consisting of Phosphate buffer (pH adjusted to 3.2 using dill. Ortho phosphoric acid) and acetonitrile, Methanol in ratio of 90:10:10 v/v and Acetonitrile ,Water in the ratio of 90:10 was selected which gave gradient elution of sharp peak with retention time at 5.954 min. Similarly for the selection of diluent we tried the standard into different solvents like water, methanol, mobile phase and acetonitrile . Finally the selected diluent was Methanol. So finally the above said mobile phase-A ,Mobile Phase-B and diluent was selected for analysis. Optimized chromatographic conditions are shown in Table 1.14-17
Table 1: Parameter Chromatographic conditions
|
Instrument
|
Water alliance High Pressure Liquid Chromatography with 2489 UV Detector |
|
Column |
Waters Symmetry C8,150x 4.6mm,3.5µm |
|
Detector |
UV detector |
|
Mobile phase-A & Mobile phase-B(Gradient Elution) |
A mixture of Phosphate buffer (pH adjusted to 3.2 using dill.Ortho phosphoric acid) and Acetonitrile , Methanol in ratio of 80:10:10v/v & Acetonitrile ,Water in the ratio of 90:10 as mobile phase-B |
|
Flow rate |
1 mL/min with Gradient Elution . |
|
Detection wave length By UV |
At 263 nm. |
|
Run time & Stop time |
40Minutes |
|
Temperature Ambient temperature |
27°C |
|
Volume of injection loop |
10μL |
|
Retention time (Rt) |
5.954 minutes |
Selection of detection wavelength
For the selection of analytical wavelength 1mg/ml Bromofenac sodium sesquihydrate solution was prepared from standard drug solution and scanned in the range of 198 to 400 nm. From the UV spectra, the maximum λmax of Bromofenac sodium sesquihydrate is found to be 263 nm. So this wavelength was selected as the detection wavelength for analysis. The selected mobile phase, diluent & wave length has given a sharp peak with low tailing factor 1.18 (<2) .
Instrumentation and analytical chromatographic conditions
The chromatographic analysis of method validation for related substance by High Pressure liquid chromatography determination of Bromofenac sodium sesquihydrate was carried out on Waters alliance High Pressure Liquid Chromatography Model -2690 series containing quaternary pump, variable wave length programmable of 2489 ultra violet visible detector and auto injector with up to 1μl-1000μl loop, column oven modules . Chromatographic analysis was performed using Waters Symmetry C8 column ,with 150 x 4.6mm internal diameter and 3.5µm particle size. Sartorius electronic balance was used for weighing . The elution was carried out Gradient at flow rate of 1 ml/min using the mixture of Phosphate buffer (pH adjusted to 3.2 using dill.Ortho phosphoric acid) and Acetonitrile , Methanol in ratio of 80:10:10v/v & Acetonitrile ,Water in the ratio of 90:10 as mobile phase-B was selected as mobile phases A and B and injection Volume 10μL(μL –micro.litre) . The detection wavelength was set at 263 nm with a runtime of 40min . The mobile phases A and B was prepared freshly and it was degassed by sonication for 5 min before use . The column was equilibrated for at least 30 min with the mobile phases A and B flowing through the system. The column oven module and the High pressure liquid chromatography system were kept at 27ºC±2°C temperature and Water: Acetonitrile and Methanol in the ratio of 33:33:33 v/v is used as rinsing solvent .
Mobile phase preparation
Mobile Phase –A
Accurately weighed 1.360 g of Potassium dihydrogen phosphate ( KH2PO4) dissolved in 1000 ml of Mili-Q-water to get phosphate buffer. pH was adjusted to 3.2±0.05 with dilute ortho phosphoric acid. Above prepared buffer and acetonitrile , Methanol were mixed in the proportion of 80:10:10 v/v. This mixture was sonicated for 10 minutes and filtered through 0.22 μm membrane filter and used as mobile phase-A .
Mobile Phase –B
Mixed Acetonitrile and Water in the proportion of 90:10 v/v , this mixture was sonicated for 10 minutes and filtered through 0.22 μm membrane filter and used as mobile phase-B .
Preparation of standard solutions and system suitability solution
Pure standards of Bromofenac sodium sesquihydrate were used as external standards in the analysis. Different concentrations of the standards were used based on the range required to plot a suitable calibration curve. About 10mg of the standard Bromofenac sodium sesquihydrate was accurately weighed and transferred in to 10 ml volumetric flask and make up with sufficient diluent. Volumetric flask containing standard solution was sonicated for 10minutes. Similarly different concentrations of these standards were analysed using the same chromatographic conditions and a calibration curve was generated. The sample chromatogram and results recorded is in Fig. 2, Table2.
Diluent
Used diluent (Methanol) as blank.
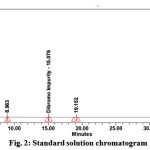 |
Figure 2: Standard solution chromatogram Click here to View Figure |
 |
Table 2: Results |
System suitability solution (or) 0.20% Impurity Blend Solution : ( Prepare fresh solution )
Weigh about 10.0 mg each of Bromofenac sodium sesquihydrate standard, Bro-4, Bro-2and Bro-3 , Dibromo Impurity in a 100 ml volumetric flask. Dissolve it by sonication till the solution is clear and make up to the mark with diluent. Dilute 200μL of the solution in to 10 ml volumetric flask and dilute up to the mark with diluent. System suitability solution or Impurity blend Solution were analysed using the same chromatographic conditions. The sample chromatogram and results recorded is in Fig. 3,Table 3.
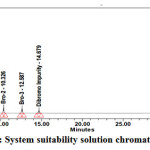 |
Figure 3: System suitability solution chromatogram |
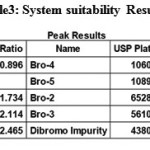 |
Table 3: System suitability Results |
Preparation of Sample solutions
Weigh accurately about 10 mg of Bromofenac sodium sesquihydrate sample to be analysed in 10 ml volumetric flask. Dissolve with about 5 ml of diluent by sonication till the solution is clear and make up to the mark with diluent.. These solutions were analysed using the same chromatographic conditions. The sample chromatogram and results recorded is in Fig. 4, Table.4.
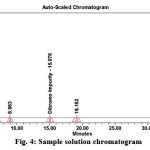 |
Figure 4: Sample solution chromatogram |
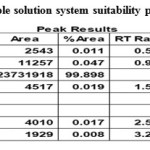 |
Table 4: Sample solution system suitability parameters |
Method Validation Procedure
After the completion of High pressure liquid chromatography method development, the objective of the method validation is to demonstrate that the method is suitable for its intended purpose as it is stated in International Conference on Harmonisation (ICH) guidelines. The method was validated for system suitability, System precision, specificity, linearity, limit of detection and limit of quantification & Range, Method precision, Robustness.18-20
System suitability Parameter
To verify that analytical system is working properly and can give accurate and precise results, the system suitability parameters are to be set. System suitability tests were carried out on freshly prepared Weigh about 10mg standard solutions of Bromofenac sodium sesquihydrate and Bro-4 in 100 ml volumetric flask. Dissolve it by sonication till the solution is clear and make up to the mark with diluent. Dilute 200 μL of the solution in to 10 ml volumetric flask and dilute up to the mark with diluent. System suitability solution were analysed using the same chromatographic conditions. it was calculated by determining the standard deviation of the values were recorded in Table 5. The system suitability method acceptance criteria set in each validation run were tailing factor ≤2.0 and theoretical plates >2000, Resolution >1.5 between two closely eluting peaks (Half width) or product specific.In all cases, the relative standard deviation (R.S.D) for the analytic peak area for two consecutive injections was not More than 2%.A chromatogram obtained from reference substance solution is presented. System suitability parameters were shown in Table.5. System suitability solution Standard chromatogram was given in Figure.5. The total results of system suitability studies summarized in Table 6. In this studies %RSD value of retention times, peak areas, tailing factor and theoretical plate count, Resolution were found to be less than 2% for Bromofenac sodium sesquihydrate (Bro-5) and 7-(4-bromobenzoyl)-1,3-dihydro-indol-2-one. [18-20].
 |
Figure 5: System suitability solution chromatogram
|
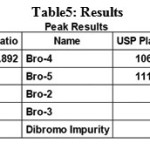 |
Table 5: Results |
Results and Discussions
System suitability parameter
Table 6: Results for System suitability parameter
|
S.no |
Name of the Impurity |
Retention Time |
Area |
Resolution |
Tailing |
Plate count |
|
1 |
BRO-4 |
5.382 |
63629 |
NA |
1.08 |
10629.82 |
|
5.385 |
63043 |
NA |
1.09 |
10663.68 |
||
|
5.389 |
63728 |
NA |
1.09 |
10656.40 |
||
|
5.386 |
62527 |
NA |
1.08 |
10630.68 |
||
|
5.374 |
62366 |
NA |
1.08 |
10659.39 |
||
|
5.305 |
62013 |
NA |
1.08 |
10686.11 |
||
|
%RSD |
0.602069 |
1.112485686 |
NA |
0.4766749 |
0.4931055 |
|
|
2 |
BRO-5 |
6.030 |
54475 |
2.93 |
1.07 |
11141.45 |
|
6.054 |
54766 |
3.02 |
1.07 |
11125.80 |
||
|
6.06 |
54536 |
3.02 |
1.06 |
11088.16 |
||
|
6.052 |
52658 |
3.04 |
1.07 |
11475.89 |
||
|
6.030 |
53396 |
2.99 |
1.07 |
11447.37 |
||
|
5.912 |
53563 |
2.9 |
1.07 |
11225.84 |
||
|
%RSD |
0.927097 |
1.528861873 |
1.8802846 |
0.3821357 |
1.508197017 |
|
System precision
The system precision is checked by using standard chemical substance to ensure that the analytical system is working properly. The retention time and area of Six determinations is measured and % RSD shall be calculated and it is Not More than 1.0%. The total results of system precision studies summarized in Table 7. [18-20].
Preparation of standard solution
Accurately weigh &transfer about 30.0mg of Bromofenac sodium sesquihydrate standard into a 10ml volumetric flask. Dissolve dilute to volume with diluent.
System precision
Table 7: System Precision parameters
| Preparation | Area |
Retention time |
Tailing Factor |
|
1 |
23731918 |
5.973 |
1.18 |
|
2 |
23531716 |
5.971 |
1.17 |
|
3 |
23631615 |
6.125 |
1.16 |
|
4 |
23431514 |
5.865 |
1.15 |
|
5 |
23519818 |
6.001 |
1.16 |
|
6 |
23481645 |
6.120 |
1.14 |
|
STDEV Average |
73982.02155 |
0.073426 |
0.014142 |
|
STDEV |
23519261.6 |
6.029167 |
1.16 |
|
%RSD |
0.31 |
1.22 |
1.22 |
From the above tabulated data, it can be concluded that the system precision parameters meets the requirements of method validation.
Specificity Parameter
Specificity is the ability of analytical method to assess the analyte in the presence of components that may be expects to be present, such as impurities, degradation products and matrix components. [18-20].
Specificity tests were carried out on above prepared standard Bromofenac sodium sesquihydrate solution of and it was determining by injecting the mobile phase and placebo solution in triplicate and recording the chromatograms specify solution (standard solution) for Bromofenac sodium sesquihydrate Standard Solution.
Table8: Specificity Parameters
|
Peak name |
RT |
|
Mobile phase |
No peaks |
|
Placebo |
No peaks |
|
Bromofenac sodium sesquihydrate standard solution |
5.985 |
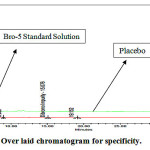 |
Figure 6: Over laid chromatogram for specificity.
|
From the above data (Table 8), (Fig. 6) Proves that method is specific that is there is no interference of placebo peaks in Bromofenac sodium sesquihydrate Standard solution.
Linearity
The linearity of an analytical method is its ability to elicit test results that are directly or by a well-defined mathematical transformation, proportional to the concentration of analyte in sample within a given range . [18-20].
The developed method has been validated as per International Conference on Harmonisation (ICH) guidelines the Standard test solutions of Bromofenac sodium sesquihydrate in the mass concentration range of 25% to 150% was injected into the chromatographic system. The chromatograms were developed and the peak area was determined for each concentration of the drug solution. Calibration curve of Bromofenac sodium sesquihydrate was obtained by plotting the peak area ratio versus the applied concentrations of Bromofenac sodium sesquihydrate. The linear correlation coefficient was found to be 1.0 (0.999). The Values & Calibration curve were recorded in Table 10 & Fig. 9. [18-20].
Linearity of Solutions from 25% to 150% was prepared with respect to the maximum percentage of known impurity level.
Preparation of linearity solutions
Table 9: Linearity different levels of concentrations
|
Level |
Weight of test solution taken with Respect to Maximum impurity level |
Dissolved and made up volume with diluent |
|
25% |
37.5mg |
100ml |
|
50% |
75.05mg |
100ml |
|
75% |
112.5mg |
100ml |
|
100% |
150.1mg |
100ml |
|
125% |
187.6mg |
100ml |
|
150% |
225.1mg |
100ml |
Injected each solution once into the HPLC system and plotted the calibration curve by taking concentration (mg/ml) on X-axis and peak area on Y-Axis and calculated the correlation coefficient .
Table 10: Linearity parameters
|
Level |
Concentration in mg/ml |
Area |
|
25% |
0.3750 |
8433 |
|
50% |
0.7505 |
16378 |
|
75% |
1.1250 |
26131 |
|
100% |
1.5010 |
35600 |
|
125% |
1.8760 |
42840 |
|
150% |
2.251 |
50767 |
|
Slope |
343.4571 |
|
|
Correlation Co-efficient |
0.999 |
|
|
Regression Coefficient |
0.998 |
|
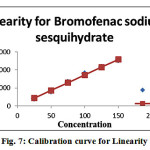 |
Figure 7: Calibration curve for Linearity |
Table 11: Residual output for Linearity parameters
|
RESIDUAL OUTPUT |
||
|
Observation |
Predicted Area |
Residuals |
|
1 |
8558.76 |
-125.7619047 |
|
2 |
17145.19 |
-767.1904762 |
|
3 |
25731.62 |
399.3809524 |
|
4 |
34318.05 |
1281.952381 |
|
5 |
42904.48 |
-64.47619048 |
|
6 |
51490.90 |
-723.9047619 |
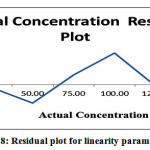 |
Figure 8: Residual plot for linearity parameters |
From the above data, it is clear that the area response of Bromofenac sodium sesquihydrate vs concentration in percentage of Bromofenac sodium sesquihydrate linear in the range of interest. The correlation coefficient and regression coefficient calculated from regular plot is greater than 0.999. Hence the method is linear for the residual determination of Bromofenac sodium sesquihydrate.
Limit of Detection and Limit of Quantification
Limit of detection is the lowest amount of analyte in a sample that can be detected, but not necessarily quantitated, under the stated experimental conditions. [18-20].
Limit of quantification is the lowest amount of analyte in a sample that can be quantitated with acceptable precision, under the stated experimental conditions. [18-20].
Limit of detection and Limit of quantitation were calculated using following formula.
LOD = (3.3 X Residual standard deviation) / slope.
LOQ = (10X Residual standard deviation) / slope .
The LOD and LOQ valuesare presented in Table 12.
Table 12: LOD & LOQ Theoretical Results
|
Levetiracetam |
|
|
Theoretical LOD in mg/ml |
0.0736mg/ml |
|
Theoretical LOQ in mg/ml |
0.2230mg/ml |
Performed a regression analysis of the linearity data with concentration vs ppm on X-axis. Calculated the residual standard deviation of the Y data. Calculated the slope of the linearity curve generated with concentration on X-axis and area response on Y-axis. The % RSD for area response of Bromofenac sodium sesquihydrate six replicates at LOQ level was found to be 0.10%.
Method Precision
Precision is a measure of the degree of repeatability of the analytical method , determined by analyzing sufficient number of aliquots of a homogenous sample solution. To study precision, three triplicate solutions of 100% and 150% Bromofenac sodium sesquihydrate were prepared and analyzed using the proposed method i.e Injected each solution once in to the chromatograph. Calculated %RSD for area. The percentage of relative standard deviation (% RSD) for peak responses was calculated and it was found to be 0.10 & 0.19% which is well within the acceptance criteria of not more than 2.0% Results of Method precision studies are shown in Table.15. [18-20].
Preparation of Method precision solutions for test solution:
Preparation of 100% test solution
Table 13: Preparations of 100% test solution
|
Spiked Prep. |
Weight taken |
Dissolved in diluent |
|
1 |
150.1mg |
100ml |
|
2 |
149.9mg |
100ml |
|
3 |
150.5mg |
100ml |
|
4 |
149.8mg |
100ml |
|
5 |
149.9mg |
100ml |
|
6 |
150.2mg |
100ml |
Preparation of 150% test solution
Table 14: Preparations of 150% test solution
|
Spiked Prep. |
Weight taken |
Dissolved in diluent |
|
1 |
225.1mg |
100ml |
|
2 |
225.2mg |
100ml |
|
3 |
225.4mg |
100ml |
|
4 |
225.3mg |
100ml |
|
5 |
224.9mg |
100ml |
|
6 |
225.5mg |
100ml |
Results of Method precision
Table 15: Method precision Results For Peak Responses of 100% and 150% of specification level
| Preparation | 100% | 150% |
| 1 | 35693 | 50767 |
| 2 | 35826 | 50787 |
| 3 | 35888 | 50757 |
| 4 | 35798 | 50747 |
| 5 | 35853 | 50748 |
| 6 | 35805 | 50999 |
| Average | 35834 | 508000 |
| STDEV | 37.00676 | 98.20064 |
| % RSD | 0.10 | 0.19 |
| Acceptance criteria | The %RSD for impurity area-Not more than 2% | |
Range
Range is defined as the range of concentration in which method is linear, precise and accurate .For range,data was considered from linearity and precise sections. Range was performed for the test solution at 25% to 150% of specification level and found it to be precise , accurate and linear. [18-20].
Linearity results
Table 16: Linearity Results for Range
| Linearity* | Correlation:0.999 |
| Acceptance criteria | Correlation Coefficient-Not less than 0.999 |
Precision results
Table 17: Precision Results for Range
| Precision(%RSD)* | |
| At 100% level | 0.10% |
| At 150% level | 0.19% |
| Acceptance criteria | The % RSD for impurity content-Not more than than 2.0% |
Test Solution and Mobile Phase Stability
Established the stability of standard solution,test solution and mobile phase which was used in estimation of % of purity, over a period of 2 days. Prepared the standard solution and test solution at 100% of specification. Prepared the mobile phase as per the test method and kept it well-closed condition. Injected blank, Standard solution and test solution freshly and injected into HPLC system by following the conditions described in test method. Calculated the % purity for test solution as per the test method. Stored the mobile phase on bench top .Stored the standard solution and test sample solution on bench top. [18-20].
On day 1 and day 2, used the stored mobile phase and injected stored system suitability solution and test solutions followed by injected freshly prepared standard solution and test solution. Test solution and mobile phase are found to be stable for 48hours from the time of preparation .System suitability results of resolution solution are within the acceptance criteria up to 48hours from the time of preparation.The results from these studies indicated, the standard & sample solution was stable at room temperature for at least 48(48h). Calculated the % of purity for stored test solutions and freshly prepared solutions as per the test method for estimation of test sample solution and mobile phase stability. Results of test solution and mobile phase stability are shown in Table.25 & 26 .The sampled chromatograms are recorded as below in Fig.11, Fig.12, Fig.13, Fig.14, Fig.15, Fig.16, Fig.17, Fig.18 & Fig.19
Solution Stability resultsand Mobile Phase Stability results
Table 18: Solution Stability result
|
Impurity |
Solution Stability |
Variation |
%Variation |
Acceptance Criteria |
||||
| Initial | 1stday | 2ndday | 1stday | 2ndday | 1stday | 2ndday | Within±15% | |
| Purity | 99.898 | 99.890 | 99.895 | 0.008 | 0.003 | 0.00001 | 0.00003 | |
Table 19: Mobile Phase Stability results
|
Impurity |
Solution Stability |
Variation |
%Variation |
Acceptance Criteria |
||||
| Initial | 1stday | 2ndday | 1stday | 2ndday | 1stday | 2ndday |
Within±15% |
|
| Purity | 99.898 | 99.889 | 99.897 | 0.009 | 0.001 | 0.0001 | 0.00001 | |
 |
Figure 9: Blank Solution Initially prepared |
 |
Figure 10: System Suitability Solution Initially prepared
|
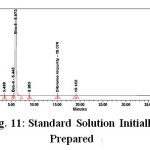 |
Figure 11: Standard Solution Initially Prepared Click here to View Figure |
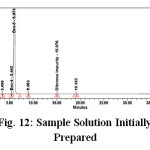 |
Figure 12: Sample Solution Initially Prepared |
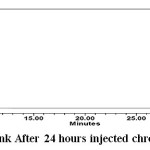 |
Figure 13: Blank After 24 hours injected chromatogram
|
 |
Figure 14: System Suitability Solution After 24 hours injected chromatogram
|
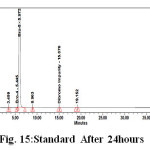 |
Figure 15: Standard After 24 hours |
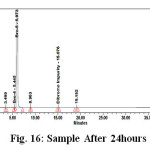 |
Figure 16: Sample After 24hours |
 |
Figure 17: Blank After 48 hours injected chromatogram
|
Robustness
Typical variations in liquid chromatography conditions were used to evaluate the robustness of the assay method. In this study, the chromatographic parameters monitored were retention time, area, capacity factor, tailing factor and theoretical plates. The robustness acceptance criteria set in the validation were the same established on system suitability test describe above. [18-20].
Table: 20
|
S.NO |
PARAMETER |
CONDITION |
AREA |
|
1 |
Standard |
Standard conditions |
23731918 |
|
2 |
Mobile phase –B |
Acetonitrile85% & Water15% |
23741028 |
|
3 |
Mobile phase |
pH 5.0 |
23751214 |
|
4 |
Wavelength |
261 nm |
23781354 |
Record of analysis for levetiracetam samples
Triplicate Bromofenac sodium sesquihydrate samples are run successfully by using this method and the experimental results and chromatograms are recorded in (Fig. 23), (Fig. 24), (Fig. 25), (Fig. 26) and (Fig. 27). [18-20].
 |
Figure 18: Blank |
 |
Figure 19: Standard chromatogram |
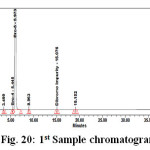 |
Figure 20: 1st Sample chromatogram |
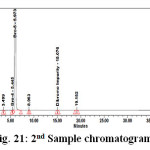 |
Figure 21: 2nd Sample chromatogram |
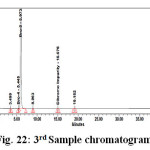 |
Figure 22: 3rd Sample chromatogram |
Significance of the developed method
Developed Gradient High Performance Liquid Chromatographic indicating method has many advantages over reported methods: (a) the method was simple because mobile phase used was cheap and easily available; (b) total run of chromatogram was 40min and Bromofenac sodium sesquihydrate (Bro-5),7-(4-bromobenzoyl)-1,3-dihydro-indol-2-one(Bro-4), (4-Bromo phenyl)-(1H-indol-7-yl) methanone (Bro-2),(3-Bromo-1H-indol-7-yl)-(4-bromo-phenyl)-methanone(Bro-3) & (4-Bromo-Phenyl)-(2,3-dibromo-1H-indol-7-yl)-methanone (Dibromo Impurity) were eluted within 20 min indicating that very less amount of mobile phase was consumed. (c) the limit of detection for Bromofenac sodium sesquihydrate was 0.0736mg/ml and the limit of quantification was0.2230mg/ml, respectively, indicating that the method was sensitive and rapid; (d) specificity study(Figs. 1 and 2) and indicate that the method was very specific, stable in the proposed method.(e) mode of separation is Gradient which mean sit is easy to operate throughout the process with out any complications (f) simultaneous estimation of different brands gives precise results indicating that developed method is compatible to estimate in different active pharmaceuticals ingradient (g) validation of the developed method as per ICH guideline indicates that the method was highly precise, rapid, simple, economical, sensitive accurate, robust and specific for determination of related impurities of Bromofenac sodium sesquihydrate in bulk and pharmaceutical dosage form.
Results
The primary target in developing & Validate Gradient Related Substance High Performance Liquid Chromatographic method developed for the analysis of Bromofenac sodium sesquihydrate in their pharmaceutical preparations method is to achieve the optimum resolution between products with other products to supply high purity of drug i.e. Based on the above observed results the developed Rapid Resolution High Performance Liquid Chromatographic (RRHPLC) validation method for Levetiracetam is valid and run successfully the summary and evaluation of results are in below (Table 28)
Table 21: Summary and Evaluation of Results
|
Validation parameter |
Acceptance criteria |
Results |
|
System suitability |
The system suitability method acceptance criteria set in each validation run were tailing factor ≤2.0 and theoretical plates >2000, Resolution >1.5 between two closely eluting peaks (Half width) or product specific.In all cases, the relative standard deviation (R.S.D) for the analytic peak area for two consecutive injections was not More than 2%. |
System suitability parameter meets the criteria and %RSD value of retention times, peak areas, tailing factor and theoretical plate count, Resolution were found to be less than 2%. |
|
System precision |
The % RSD of retention time and area of 10 determinations should not more than 1.0% |
System precision parameters meets the requirements of method validation. |
|
Specificity |
No interference of placebo peaks in Bromofenac sodium sesquihydrate Standard solution |
Method is specific that there is no interference of placebo peaks in Bromofenac sodium sesquihydrate Standard solution |
|
Linearity |
The correlation coefficient and the regression coefficient between concentration and area response of Bromofenac sodium sesquihydrate should be NLT 0.999 |
The correlation coefficient and regression coefficient calculated from regular plot is greater than 0.999 |
|
LOD/LOQ |
The% of RSD for area response of Bromofenac sodium sesquihydrate from six replicates at LOQ level should be NMT 10.0% |
LOD/LOQ parameters meets the requirements of method validation. |
|
Method Precision |
% of RSD for purity and Area at 100% of specification and 150% of specification levet not more than 2.0% |
Method precision parameter meets the criteria and %RSD value of retention times, peak areas were found to be less than 2%. |
|
Range
|
At 25% to 150% of specification level to be precise , accurate and linear. |
Meet the criteria At 25% to 150% of specification level and found it to be precise , accurate and linear. |
|
Test solution and Mobile phase Stability |
Established the stability of standard solution ,test solution and mobile phase which was used in estimation of % of purity, over a period of 2 days. |
The results from these studies indicated, the standard & sample solution , Mobile phase was stable at room temperature for at least 48(48h). |
|
Robustness |
A small deliberate variations in method parameters like Mobile Phase –B (± 5% of organic Phase),Wave length (± 2nm), and pH (± 0.2). |
From the results reveal that the method is robust. |
Conclusion
The Gradient Related Substance High Performance Liquid Chromatographic method developed for the analysis of Bromofenac sodium sesquihydrate in their pharmaceutical preparations is precise, accurate, and with a reasonable run time. The developed method was validated as per ICH Guidelines shows that the developed method was highly specific and robust so that it can be effec-tively applied for routine analysis in research institutions, in quality control department of pharmaceuticals industries, and in approved testing laboratories, from the above experimental data on the various method validation parameters, it is proved that this method which was designed to determine the related impurities in Bromofenac sodium sesquihydrate is precise, accurate, linear, rugged, robust and range from 25% to 150% of the Specification . Hence, the method can be used for routine application.
References
- Sancilio L F, Nolan J C, Wagner L E, Ward J W. “The analgesic and anti-inflammatory activity and pharmacologic properties of bromfenac. Arzneimittelforschung.” 1987, 37(5), 513–519. [PubMed].
- Ruiz J, Lopez M, Mila J, Lozoya E, Lozano J J, Pouplana R., “QSAR and conformational analysis of the anti-inflammatory agent amfenac and analogues”. J Comput Aided Mol Des. 1993, 7(2), 183–198. [PubMed].
- Guex-Crosier Y. “Non-steroidal anti-inflammatory drugs and ocular inflammation. Klin Monbl Augenheilkd.” 2001, 218(5), 305–308. [PubMed].
- O’Banion M K, Winn V D, Young D A. “CDNA cloning and functional activity of a glucocorticoid-regulated inflammatory cyclooxygenase.” Proc Natl Acad Sci USA. 1992, 89(11), 4888–4892. [PubMed].
- Waterbury L D, Silliman D, Jolas T. “Comparison of cyclooxygenase inhibitory activity and ocular anti-inflammatory effects of ketorolac tromethamine and bromfenac sodium.” Curr. Med Res Opin., 2006, 22(6), 1133–1140. [PubMed].
CrossRef - Xibrom (bromfenac ophthalmic solution) 0.09% [package insert full prescribing information] Irvine, CA: Ista Pharmaceuticals; 2008.
- United States of Pharmacopoeia, United StatesPharmacopoeia Convention, Rockville.2009; 1328.
- Bucci F A, Jr Waterbury L D. “Comparison of ketorolac 0.4% and bromfenac 0.09% at trough dosing: aqueous drug absorption and prostaglandin E2 levels.” J Cataract Refract Surg. 2008, 34(9), 1509–1512. [PubMed]
- Bronuck [package insert full prescribing information] Osaka, Japan: Senju Pharmaceutical Company, Ltd; 2003.
- Bromday (bromfenac ophthalmic solution) 0.09% [package insert full prescribing information] Irvine CA: Ista Pharmaceuticals, 2010.
- Yellox (bromfenac sodium sesquihydrate) [package insert full prescribing information] Leobendorf, Austria: Croma Pharma GmbH; 2011.
- European Pharmacopoeia, Strasbourg Supplement, 5th ed., Coun-cil of Europe, 2005, 2925, 3440
- British Pharmacopoeia, British Pharmacopoeia Commission,TSO, London, 2007, 140, 876.
- R. Snyder, J. Kirkland, L. Glajch. Practical HPLC method development. II Ed. A Wiley International publication;1997., 235,266-268,351-353.653-600.686-695.
- Chen Y., Hua T., “Related substances by ion-pairs along with gradient elution HPLC method”, Chin. J. Antibiot. 2004, 29 15–18 .
- Skoog D A, Holler J, Nieman T A; “Principle of Instrumental Analysis.5th Edition “ . 778-787.
- Sharma B K “Instrumental Methods of Chemical Analysis. GOEL Publication House. Meerut.”.133-161, 68-80, 114-165, 286-320.
- “ICH Q2 (R1) Validation of analytical procedures”, Text and methodology International Conference on Harmonization, Geneva. 2005; 1-17.
- “Validation of Analytical Procedures: Q2A, Definitions and Terminology”, ICH, October 1994; Q2B, Methodology, ICH, November 1996.
- “Guideline in sterile drug products produced by aseptic processing Center for Drug Evaluation and Research Food and Drug Administration” June 1987.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


