Virtual Screening of Natural Products, Molecular Docking and Dynamics Simulations on M.tuberculosis S-adenosyl-L-homocysteine Hydrolase
Abdul-Rashid B. Sampaco Iii1 and Junie B. Billones1,2*
1Department of Physical Sciences and Mathematics, College of Arts and Sciences, University of the Philippines Manila, Padre Faura, Ermita, Manila, 1000 Philippines 2Institute of Pharmaceutical Sciences, National Institutes of Health, University of the Philippines, Pedro Gil, Ermita, Manila 1000, Philippines. Corresponding Author Email :jbbillones@up.edu.ph
DOI : http://dx.doi.org/10.13005/ojc/310402
Article Received on :
Article Accepted on :
Article Published : 10 Nov 2015
The activated methyl cycle of Mycobacterium tuberculosis(Mtb)is responsible for the regeneration of S-adenosyl methionine (SAM) from S-adenosyl-L-homocysteine (SAH). Inhibition of the key enzymes in this transformation may lead to accumulation of SAH and depletion of SAM in the Mtb cell. This has detrimental effects onthe bacterium’s cellular processes. Virtual screening of natural products from the Philippines and those in Ambinter database against S-adenosyl-L-homocysteine hydrolase (SAHH) yielded the tautomer of the molecule, methyl 4-({2-[(4-hydroxy-2-oxo-1,2-dihydro-3-quinolinyl)carbonyl]hydrazino}sulfonyl)phenylcarbamate, which displays better binding energy (-307.64 kcal/mol) than the substrate, SAH (-270.601 kcal/mol). Molecular dynamics simulations at body temperature indicated that the hit-SAHH complex is more stable than the enzyme-substrate complex.
KEYWORDS:Mycobacterium tuberculosis; S-adenosyl methionine (SAM); S-adenosyl-L-homocysteine (SAH); virtual screening; molecular docking; molecular dynamics; phenylcarbamate
Download this article as:| Copy the following to cite this article: Iii A. B. S, Billones J. B. Virtual Screening of Natural Products, Molecular Docking and Dynamics Simulations on M.tuberculosis S-adenosyl-L-homocysteine Hydrolase. Orient J Chem 2015;31(4). |
| Copy the following to cite this URL: Iii A. B. S, Billones J. B. Virtual Screening of Natural Products, Molecular Docking and Dynamics Simulations on M.tuberculosis S-adenosyl-L-homocysteine Hydrolase. Orient J Chem 2015;31(4). Available from: http://www.orientjchem.org/?p=12550 |
Introduction
Tuberculosis (TB), one of the world’s most deadly infectious diseases, is caused by the bacterium called Mycobacterium tuberculosis(Mtb). According to the World Health Organization (WHO), Mtb is ranked second to HIV/AIDS as the deadliest single infectious agent worldwide. In 2013, approximately 9.0 million people developed TB, which caused 1.5 million deaths in the same year. 1 The current treatment for TB consists of the administration of four first-line drugs – isoniazid, rifampicin, ethambutol, pyrazinamide – for two months, followed by 4 months of treatment with isoniazid and rifampicin.1This treatment however, is too time-consuming and is not usually compatible with medication for treatment of other conditions like HIV. Improper and immature use and monitoring of the drug regimen have led to the emergence of multidrug resistant, extensively drug resistant, and totally drug resistant TB strains.2 – 4
Part of discovering new drugs to combat TB is by targeting novel enzymes. One of the new attractive drug targets in Mtb is S-adenosyl-L-homocysteine hydrolase (SAHH), an enzyme in the activated methyl cycle. The activated methyl cycle is responsible for the regeneration of S-adenosyl methionine (SAM) from S-adenosyl-L-homocysteine (SAH).5Compounds that hamper the activated methyl cycle cause the accumulation of SAH and depletionof SAM.SAM, the end product of the cycle, is a donor of active methyl groups in several essential cellular reactions and also a cofactor of certain enzymes.6Particular ratio of SAM to SAH in bacterial cell should be maintained for survival, and perturbation of this ratio level leads to growth arrest.7The processes that require methyl groups from SAM include DNA methylation, polyamine and N-acylhomoserine lactone biosyntheses, biotin formation,8 and ribosomal RNA methylation.9,10In M. tuberculosis, the SAM-dependent methyltransferases include Hma11, which catalyzes keto and methoxy-mycolic acids, and Rv2952.12Another SAM dependent methyltransferase in Mtb encoded by umaA catalyzes the conversion of the direct methylation ofphospholipid-linked esterified oleic acid to essential fatty acid, 10-methylstearic acid(or Tuberculostearic acid, TSA).13
Since in vitro and in vivo testing of binding affinities of a vast collection of compounds to TB protein targets are time consuming and expensive, computer-aided drug discovery has been used as a more practical and efficient alternative for speedy identification of potential leads. This study has been focused on SAHH as thedrug target, based on which a pharmacophore was developed and used to screen a database of natural products. The interaction of the natural products with SAHH was modeled using molecular docking and molecular dynamics techniques.
Experimental Section
The visualization andoptimization of protein and ligand structures, virtual screening, molecular docking,calculation of binding energies, and molecular dynamics were performed by the use of BIOVIA’s Discovery Studio® (DS) package.14
Protein Preparation and Minimization
The crystal data for S-adenosyl-L-homocysteine hydrolase were retrieved from Research Collaboration for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) (http://www.rcsb.org/). For M. tuberculosis, the protein (PDB ID: 3DHY) bound to ethylthioadenosine, a close analog of the substrate was used.6The protein structure was preparedusing the Prepare Protein protocol of DS.The output file obtained from preparation was used as the input file in structure optimization using the Minimization protocol of DS. The resulting prepared and minimized proteinstructure was used for all other subsequent protocols.
Site Sphere Definition
The site sphere locates and defines the binding site based onthe position of the bound ligand on the original protein crystal structure. Since the protein in thisstudy was homotetrameric, only onesite sphere was generated. Thus,only one ligand wasselected and a site sphere was generated using the Define Sphere protocol. The coordinates of the site sphere were used to makeidentical copies on the prepared and minimized structure.
Pharmacophore Generation
Pharmacophore features based on the active site of Mtb SAHH weregenerated using the Interaction Generation protocol. The generatedpharmacophore features were clustered together using the Cluster CurrentFeatures and Keep Clusters Only tools based on their type as acceptor,donor, and hydrophobe.
Ligand Preparation and Building of 3D Databases
The molecules docked were a compilation of 1029 Philippine natural products and the Ambinter Database (www.ambinter.com). Over 75,000 structures were prepared using the Prepare Ligands protocol. The prepared ligands weredivided into smaller groups. Each group was built into respective 3Ddata bases using the Build 3D Database protocol.
Rigid and Flexible Fitting
The virtual screening of compounds was composed of two parts: rigid and flexible fitting. After rigid fitting, all ligands with Fit Values of 2.9 or higher were forwarded to screening by flexible fitting method. All ligands with Fit Value of 3.2 or higher were then docked to the target enzyme.
Molecular Docking and Calculation of Binding Energies
The CDOCKER protocol was used for docking each hit to SAHH. Calculate Binding Energies protocol was used for computing binding affinity of each complex. The binding energy value of the SAH-SAHH complex was used as baseline.
Molecular Dynamics
Standard Dynamics Cascade protocol was used for the moleculardynamics simulations. Its output contains different conformations of theprotein at different temperatures. Three dynamics simulations were done: 1.) Mtb SAHH alone, 2.) Mtb SAHH bound to substrate SAH, and 3.)Mtb SAHH bound to the top hit. The potential energies of their conformations at the body temperature (310 K) were compared. Ligand interaction diagrams were also generated for analysis of interactions.
Results and Discussion
Preparation of the Target Protein
Out of five crystal structures of Mtb SAHH available in PDB, the one bound to ethylthioadenosine was chosen because of the close resemblance of this ligand to the substrate. Like SAH, ethylthioadenosine also possesses a ribose ring bonded to an adenine through an N-glycosidic bond. The only difference in their structure is that in ethylthioadenosine, the 5’ of the ribose ring is attached to a thioethyl group rather than homocysteine. The target protein was prepared by inserting missing main- and side-chain atoms, optimizing the conformation on the area where the missing atoms were added, and removing the water molecules. The potential energy was subsequently minimizedthrough geometry optimization of the protein structure. The potential energyof the minimized structure of Mtb SAHH was found to be -135,696 kcal/mol. Moreover, the deviation of the prepared protein structure (Figure 1) from the original crystal structure was evaluated. The two structures were overlaid using a carbon stick diagram (Figure 2)for ocular comparison. The overlay diagram shows very little deviation in their structures (RMSD = 0.762Å for the main chain atoms).
 |
Figure 1: A solid ribbon diagram of the prepared structure of Mtb SAHH. |
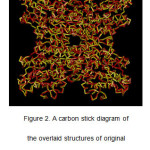 |
Figure 2: A carbon stick diagram of the overlaid structures of original crystalstructure(red) and prepared and minimized structure (yellow) of Mtb SAHH. |
SAHH is a tetramer with four identical subunits. Each subunit contains onebinding site and the residues on the binding site of onesubunit is identical to the residues on the other subunits. Experimentally, the adenine ring of SAH was surrounded by Leu68, Thr71, Gln73, Leu410, Met421, and Phe425 residues.6 The two hydroxyl groups of the ribose ring interacted with ionizablegroups of Asp156, Glu218, Lys258, and Asp252. Anotherinteraction was found between the carboxylate group and His363.
A ligandinteraction diagram for ethylthioadenosine in complex with SAHH also showed that the key amino acids at the active site also interacted with ethylthioadenosine. The only aminoacid that was not seen in the diagram is His363. This observation is justified because His363 is supposed to interact with the carboxylate group of SAH,which is absent in ethylthioadenosine.
To define and mark the binding site on the structure, a site sphere around the bound ligand was generated (Figures 3). The coordinates of the sphere in the Mtb SAHH were (19.439, -9.701,40.371) with a radius of 8.72 Å.
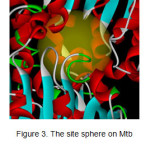 |
Figure 3: The site sphere on Mtb SAHH viewed closely. |
Ligand Preparation and Database Construction
The molecules that were screened came from Philippine natural sources and the Ambinter Natural Product Database. These two databasescontain a total of 75,702 molecules. Prior to any computational protocol, theywere first prepared by enumerating their isomers, tautomers, and ionizationstates. 3D conformations of each were then generated. After preparation, themolecules were consolidated as databases ready for screening.
Pharmacophore Generation and Virtual Screening against Mtb SAHH
A pharmacophore was generated based on the active site of Mtb SAHH, by analyzing the active site for donors, acceptors, and hydrophobes. A total of 1,775 features were generated and subsequently clustered down to only 17 features consisting of 6 donors, 7 acceptors, and 4hydrophobes (Figure 4).
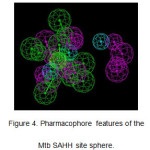 |
Figure 4: Pharmacophore features of the Mtb SAHH site sphere. Click here to View figure |
The 17-feature pharmacophore was used to screen the databases of natural products against SAHH. The firstscreening process was done through rigid fitting. The arbitrary fit value cut-off was set to 2.9. All molecules that did not reach the cut-off were eliminated. Out of theinitial 75,702 molecules that were screened, a total of 3,746 reached the cutoffand were passed on to the next step. All of these molecules were re-screened through flexible fitting method. With the arbitrary cut-off set at 3.2, only 1,509 molecules passed through the screen and were set for the subsequent molecular docking procedure.
Molecular Docking and Binding Energy Calculations
The CDOCKER docking protocol works by first generating several conformations of the ligand bycontinuously adding thermal energy. This is a molecular dynamics method that treat the ligand as a flexible structure.5 Each conformation is then directed into an area in the site sphere where it is randomly rotated several times. Each resulting orientation is saved as one pose. The program was set such that only the top ten poses in terms of binding energy are reported. The 1,509 molecules that passed both rigid and flexible fitting were docked onto the binding site of Mtb SAHH. In addition, the prepared SAH was also docked onto the protein for reference.
The binding energy of the substrate, SAH, to the protein was found to be -270.60 kcal/mol. Analysis of its ligand interaction diagram (Figure 5),revealed that all amino acid residues found in experiment were also present in the diagram. Unlike ethylthioadenosine, the carboxylate group of SAH was shown to exhibit charged interactions with His363. There are a total of four observable H-bonds andone charged interaction. His416 behaves as an H-bond donor to N-7 of adenine and an H-bond acceptor to its amino group. Side chains of Thr220 and Asp252 also behave as H-bond acceptors to the alpha amino group and aribose hydroxyl group, respectively.
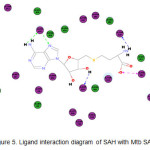 |
Figure 5: Ligand interaction diagram of SAH with Mtb SAHH. |
Out of the 1,509 molecules that were docked, only one was found to have better binding affinity to the protein than SAH. This molecule, a tautomer of methyl 4-({2-[(4-hydroxy-2-oxo-1,2-dihydro-3-quinolinyl)carbonyl]hydrazino}sulfonyl)phenylcarbamate (Figure 6), has a binding energy of -307.64kcal/mol. Figure 7 shows thatall amino acidsreported to interact with SAH, save Leu68, were also found to interact withthe top hit. It is observed that its sulfone group plays an important role in itsbinding activity. The two oxygen atoms both participate in H-bond interactions with Thr219 and Lys 248. There is also an interaction between the pi electrons of the aromatic ring with the positively charged imidazole ring of His69.
![Figure 6. Structure of the top hit, methyl 4-({2-[(4- hydroxy-2-oxo-1,2-dihydro-3-quinolinyl)carbonyl]hydrazino}sulfonyl)phenylcarbamate.](http://www.orientjchem.org/wp-content/uploads/2015/11/Vol31_No4_Vir_ABD_Fig6-150x150.jpg) |
Figure 6: Structure of the top hit, methyl 4-({2-[(4- hydroxy-2-oxo-1,2-dihydro-3 quinolinyl)carbonyl]hydrazino}sulfonyl)phenylcarbamate. |
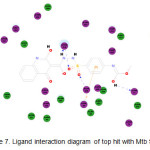 |
Figure 7: Ligand interaction diagram of top hit with Mtb SAHH. |
Molecular Dynamics
Molecular dynamics is a computational method in which the motions of amolecule brought about by a change in the environment arepredicted and simulated. In this study, the standardized dynamicsprotocol of DS was used. An input protein structure was minimized through two intensive minimization protocols to bring its energy to the minimum. Thermal energy was continually added to the minimized structure until it reached a target temperature, i.e. body temperature. The structure was then equilibrated such that the energy applied was distributed over the whole structure. In this way,the most stable conformation at a specified temperature was generated. The process was done repeatedly until 10 conformations of the protein at different temperatures ranging from 298 K to 310 K were generated. The potentialenergy of each conformation was also calculated.
The molecular dynamics simulation done on Mtb SAHH at 310K resulted in a slight deviation from the minimized structure. The RMSD calculations showed that there is a deviation of 1.066 Åon the main chain atoms and 1.024Å on the alphacarbons. Unlike molecular docking, molecular dynamics simulation does not “dock” several conformations of a molecule to a rigid protein structure. Both the protein and the ligand are treated as flexible structures. Basically, the program determines the lowest energy conformation of the complex at different temperatures.Therefore, two features of molecular dynamics explain why it is more accurate than molecular docking: 1) it treats the whole complex, both proteinand ligand, as a flexible structure, and 2) it simulates binding at several temperatures. In this study, the complexes of the protein with both the to phit and the substrate were simulated separately. Their conformations at 310 K were evaluated and their potential energies were compared. The potential energy of the protein-substrate complex was -118,996 kcal/mol while that of the protein-top hit complex was -119,099kcal/mol. This corroborates the results of the molecular docking procedure, the potential energy of the protein with the top hit being more negative than that of the protein bound to the substrate. In other words, the complex formed by the protein and the top hit is more stable than the protein-substrate complex.
The ligand interaction diagram of the SAHH-SAH complex at 310 K was compared with that obtained from molecular docking. There are some interactions that were consistent with the diagram generated from the docking simulation. These interactions are the H-bond interactions between Asp252 and a hydroxyl group of the ribose, between Thr220 and the alphaamino group, and the charged interaction between His363 and the carboxylate group. However, additional pi-pi interactions between the aromatic adenine rings and the imidazole rings of His69 and His416 were observed in dynamics simulation. The ligand interaction diagram of the SAHH-top hit complex was also compared to that generated from the docking simulation. Allnotable interactions observed in the docking simulation were also present in the dynamics simulation. However, there are additional interactions that also warrant attention. H is 416 participated in two interactions: it acts as an H-bond donor to one of the nitrogens in the hydrazine group, andexhibits a charged interaction with the oxygen of the enolate. It should alsobe noted that unlike in the docking simulation, His69 exhibitscation-pi interactions with all aromatic rings of the ligand. Lastly, there are two additional amino acids that exhibit pi interactions with the ligand’simidazole ring, Phe364 and His363.
Conclusion
Pharmacophore-based virtual screening of over 75,000 natural products from Philippine sources and Ambinter database against Mtb SAHH was carried out. Both molecular docking and dynamics simulations established the top hit, a tautomer of methyl 4-({2-[(4-hydroxy-2-oxo-1,2-dihydro-quinolinyl)carbonyl]hydrazino}sulfonyl)phenylcarbamate as a good inhibitor of SAHH and a potential lead compound against TB.
Conflict of Interest
The authors declare that there is no conflict of interest in this work.
Acknowledgement
We thank the Emerging Inter-Disciplinary Research (EIDR) program (OVPAA-EIDR 12-001-121102), OVPAA, University of the Philippines System for the use of computational facilities.
References
- WHO. World Health Organization Global Tuberculosis Report 2014, Geneva 2014. http://apps.who.int/iris/bitstream/10665/137094/1/9789241564809_eng.pdf. (Retrieved September 2015).
- Babu, G.; Laxminarayan, R.The unsurprising story of MDR-TB resistance in India.Tuberculosis, 2012, 92, 301-306.
- Gandhi, N.; Moll, A.; Sturm, A.W.; Pawinski, R.; Govener, T.; Lalloo, U.; Zeller, K.; Andrews, J.;Friedland, G.Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa.Lancet, 2006, 368(9547), 1575-1580.
- Velayati, A.A.; Farnia, P.;Masjedi, M.R.; Ibrahim, T.A.; Tabarsi, P.; Haroun, R.Z.; Kuan, H.O.; Ghanavi, J.; Farnia, P.; Varahram, M. Totally drug-resistant tuberculosis strains: evidence of adaptation at the cellular level.Eur. Respir. J.,2009, 34(5), 1202-1203.
- Schauder, S.; Shokat, K; Surette, M.G.; Bassler, B.L.The LuxS family of bacterial autoinducers: biosynthesis of a novel quorum-sensing signal molecule. Mol. Microbiol.,2001, 41(2), 463-476.
- Reddy, M.C.; Kuppan, G.; Shetty, N.D.; Owen, J.L.; Ioerger, T.R.; Sacchettini,J.C.Crystal structures of Mycobacterium tuberculosis S-adenosyl-L-homocysteine hydrolase in ternary complex with substrate and inhibitors.Protein Sci.2008, 17(12), 2134–2144.
- Kramer, D.L.; Porter, C.W.; Borchardt, R.T.;Sufrin, J.R.Combined Modulation of 5-AdenosyImethionine Biosynthesis and 5-Adenosylhomocysteine Metabolism Enhances Inhibition of Nucleic Acid Methylation and LI 210 Cell Growth.Cancer Res.,1990, 50, 3838–3842.
- Parveen, N.; Cornell, K.Methylthioadenosine/S-adenosylhomocysteine nucleosidase, a critical enzyme for bacterial metabolism. Mol. Microbiol.2011, 79(1), 7-20.
- Kumar, A; Saigal, K; Malhotra, K; Sinha, KM; Taneja, B.Structural and Functional Characterization of Rv2966c Protein Reveals an RsmD-like Methyltransferase from Mycobacterium tuberculosis and the Role of Its N-terminal Domain in Target Recognition.J. Biol. Chem., 2011, 286(22), 19652–19661.
- Rahman, A; Srivastava, SS; Sneh, A; Ahmed, N; Krishnasastry,MV.Molecular characterization of tlyA gene product, Rv1694 of Mycobacterium tuberculosis: a non-conventional hemolysin and a ribosomal RNA methyl transferase. BMC Biochem., 2010, 11, 35.
- Boissier, F; Bardou, F; Guillet, V; Uttenweiler-Joseph, S; Daffe, M; Quemard, A; Mourey, L.Further insight into S-adenosylmethionine-dependent methyltransferases: structural characterization of Hma, an enzyme essential for the biosynthesis of oxygenated mycolic acids in Mycobacterium tuberculosis. J. Biol. Chem.,2006, 281, 4434–4445.
- Huet, G; Constant, P; Malaga, W; Laneelle, MA; Kremer, K; van Soolingen, D;Daffe,M; Guilhot, C.A lipid profile typifies the Beijing strains of Mycobacterium tuberculosis.Identification of a mutation responsible for a modification of the structures of phthiocerol dimycocerosates and phenolic glycolipids.J. Biol. Chem.,2009, 284, 27101–27113.
- Meena, L.; Chopra, P; Vishwakarma, R; Singh,Y.Biochemical characterization of an S-adenosyl-l-methionine-dependent methyltransferase (Rv0469) of Mycobacterium tuberculosis. Biol. Chem. 2013, 394(7), 871–877.
- BIOVIA, 5005 Wateridge, Vista Drive, San Diego CA 92121, USA. (http://accelrys.com)
- Wu, G; Robertson, DH; Brooks, CL; Vieth,M.Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm.J. Comput. Chem.2003, 24(13), 1549-1562.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


