SWNTs and MWNTs (M=2) for Nano Drug Delivery: Temperature and Solvent effect
Elham Shabanzadeh* and Roghieh Tarlani Bashiz
Department ofChemistry, Science and Research Branch, Islamic Azad University, Tehran, Iran Corresponding Author Email: e_shabanzadeh@yahoo.com
DOI : http://dx.doi.org/10.13005/ojc/310481
Article Received on :
Article Accepted on :
Article Published : 09 Dec 2015
The drug delivery via multi wall nano tube has been studied using ab-initio and QM/MM methods. We have studied the solvent effects on the relative energies and dipole moment values and the structural properties of water, methanol and ethanol surrounding single-walled and multi walled carbon nanotube. In this study we investigated the polar solvents effects on MWCNT within the Onsager self - consistent reaction field (SCRF) model using a Hartree-Fock method and the temperature effect on the stability of SWCNT in various. Because some of the physicochemical parameters related to structural properties of SWCNT, we used different force fields to determine energy and other types of geometrical parameters, on the particular SWCNT. It is important to understand the energetic, stability dependent physical properties of armchair (m, n) carbon nanotube for specific molecules of -N=C=S group reacts.
KEYWORDS:SWCNT; DFT; Thermodynamic; NMR; MWNTs; antibiotics; -N=C=S group reacts
Download this article as:| Copy the following to cite this article: Shabanzadeh E, Bashiz R T. SWNTs and MWNTs (M=2) for Nano Drug Delivery: Temperature and Solvent effect. Orient J Chem 2015;31(4). |
| Copy the following to cite this URL: Shabanzadeh E, Bashiz R T. SWNTs and MWNTs (M=2) for Nano Drug Delivery: Temperature and Solvent effect. Orient J Chem 2015;31(4). Available from: http://www.orientjchem.org/?p=13204 |
Introduction
The highly electrophilic central carbon atom of the -N=C=S group reacts rapidly, and under mild conditions with oxygen-, sulfur-, or nitrogen centered nucleophiles to give rise to carbamates, thiocarbamates, or thiourea derivatives, respectively. The -NCS group of isothiocyanates absorbs UV light with low intensity near 240 nm. Among the most extensively investigated isothiocyanates is PEITC which occurs naturally as glucosinolate in a variety of cruciferous vegetables such as: kale, turnip, cabbage and broccoli. Phenethyl-ITC (PEITC) is one of the best-studied members of the ITC family of compounds that has generated a great deal of research interest due to its cancer chemopreventive activity [1-10]. Angiogenesis (formation of new blood vessels): cancer, rheumatoid arthritis, endometriosis and diabetic retinopathy. Therefore, antiangiogenic therapy represents one of the most promising approaches to control tumor growth and invasiveness. PEITC inhibits transcriptional activity of nuclear factor-kB (NF-kB) and suppresses expression of NF-kB–regulated genes, including vascular endothelial growth factor (VEGF) 3, 4], Because VEGF plays an important role in angiogenesis by promoting endothelial cell proliferation, migration, and differentiation. PEITC effectively inhibits in vitro angiogenic features. PEITC effectively inhibited chemically-induced lung, mammary gland, forestomach and esophagus tumorigenesis. Phenethylisothiocyanate exhibits antileukemic activity in vitro and in vivo [2-10].
The carbon nanotube (CNT) is a representative nano-material. CNT is a cylindrically shaped carbon material with a nanometric-level diameter [11-18].
Its structure, which is in the form of a hexagonal mesh, resembles a graphite sheet and it carries a carbon atom located on the vertex of each mesh. The sheet has rolled and its two edges have connected seamlessly [19-25].
Although it is a commonplace material using in pencil leads, its unique structure causes it to present characteristics that had not found with any other materials. CNT can be classified into single-wall CNT, double-wall CNT and multi-wall CNT according to the number of layers of the rolled graphite [26,30].
The type attracting most attention is the single-wall CNT, which has a diameter deserving the name of “nanotube” of 0.4 to 2 nanometers. The length is usually in the order of microns, but single-wall CNT with a length in the order of centimeters has recently released [31,40].
CNT can be classified into single-wall CNT, double-wall CNT and multi-wall CNT according to the number of layers of the rolled graphite. The type attracting most attention is the single-wall CNT, which has a diameter deserving the name of “nanotube” of 0.4 to 2 nanometers [28-32].
The length is usually in the order of microns, but single-wall CNT with a length about centimeters have recently released. The extremities of the CNT have usually closed with lids of the graphite sheet [40-49].
The lids consist of hexagonal crystalline structures (six-membered ring structures) and a total of six pentagonal structures (five-membered ring structures) placed here and there in the hexagonal structure [50-55]. The first report by Iijima was on the multiwall form, coaxial carbon cylinders with a few tens of nanometers in outer diameter [49-56]. Two years later single walled nanotubes were reported [57-63]. SWCNTs have considered as the leading candidate for nano device applications because of their one-dimensional electronic bond structure, molecular size, and biocompatibility, controllable property of conducting electrical current and reversible response to biological reagents hence SWCNTs make possible bonding to polymers and biological systems such as DNA and carbohydrates[64-69].
So, The structure ofSWCNT as well as their dipole moments and relative energies have been studied by molecular dynamics simulation and quantum mechanics calculations within the Onsager self – consistent reaction field (SCRF) model using a Hartree-Fockmethod (HF) at the HF/3-21G level and the structural stability of considered nanotube in different solvent media and temperature have been compared and analyzed.We investigate effects of water, methanol and a mixture of them on interaction of Temozolomide with SWCNT, utilizing these force fields too [48-66].
The calculations have been done with the GAUSSIAN 98 program according to Hartree-Fock (HF) theory at the HF/STO-3G level. Gibbs free energy,enthalpy,entropy and dipole moment values are compared in gas phase, water and methanol, in this research[64-80]. Thus, by utilizing a Hartree-Fock method, we studied the effects of different solvents on interaction of Temozolomide with open-end of SWCNT within the onsager self-consistent reaction field (SCRF) model, and the temperature effects on the stability of the interaction between Temozolomide and single-walled carbon in various solvents[80-97].
Computional Details
The term “Ab Initio” is given to computations which are derived directly from theoretical principles, with no inclusion of experimental data. The most common type of ab initio calculation is called a HartreeFock calculation (abbreviated HF), in which the primary approximation is called the central field approximation. A method, which avoids making the HF mistakes in the first place, is called Quantum Monte Carlo (QMC). There are several flavors of QMC variational, diffusion and Green’s functions. These methods work with an explicitly correlated wave function and evaluate integrals numerically using a Monte Carlo integration. These calculations can be very time consuming, but they are probably the most accurate methods known today. In general, ab initio calculations give very good qualitative results and can give increasingly accurate quantitative results as the molecules in question become smaller.
There are three steps in carrying out any quantum mechanical calculation in HyperChem 7.0 program package [68]. First, prepare a molecule with an appropriate starting geometry. Second, choose a calculation method and its associated options. Third, choose the type of calculation with the relevant options.DFT is based on a theorem due to Hohenberg and Kohn, which states that all ground state properties are functions of the total electronic charge density ρ(r).There are several different DFT functional available differing primarily in the choice of the basis functions, in which, the electronic wave functions are expanded and the scheme of integration.[69,70].
The Becke’s three parameter exact exchange functional (B3) combined with gradient corrected correlation functional of Lee–Yang–Parr (LYP) have been employed to calculate energy, dipole moment, charge distribution and thermo chemical data and NMR parameters by implementing the 6-31G,STO-3G basis sets. All the NMR shielding parameters were calculated supposing gauge-included atomic orbital (GIAO) method.
NMR spectroscopy is a research technique that exploits the magnetic properties of certain atomic nuclei to determine physical and chemical properties of atoms or the molecules in which they are contained. It relies on the phenomenon of nuclear magnetic resonance and can provide detailed information about the structure, dynamics, reaction state, and chemical environment of molecules. Ab initio calculation of nuclear magnetic shielding has become an indispensable aid in the analysis of molecular structure and accurate assignment of NMR spectra of compounds.
NMR is based on the quantum mechanical property of nuclei. The chemical shielding refers to the phenomenon, which is associated with the secondary magnetic field created by the induced motions of the electrons that surrounding the nuclei when in the presence of an applied magnetic field. In general, the electron distribution around a nucleus in a molecule is more spherically symmetric. Therefore, the size of electron current around the field, and hence the size of the shielding, will depend on the orientation of the molecule within the applied field B0.
Calculations of nucleus-dependent and -independent chemical shifts were carried out using the gauge-invariant atomic orbital (GIAO) approach.
Calculations were performed using an all-electron linear combination of atomic orbitals Hatree–Fock (HF) and density functional theory (DFT) calculations using the Gaussian 03 package. The optimizations of (2-fluoro-2-isothiocyanatoethyl)benzene, (2-chloro-2 isothiocyanatoethyl)benzene, (2-bromo-2-isothiocyanatoethyl)benzene, (2 isothiocyanatopropyl)benzene, (2-isothiocyanato Butyl)benzene and (1-isothiocyanatoethane-1,2-diyl) di benzene (used in this study are shown in Figure. 2) are carried out including exchange and correlation contributions using Beckẻs three parameter hybrid and Lee-Yang-Parr (LYP) correlation [B3LYP]; including both local and non-local terms. We have geometric optimization calculation at the HF/6-31G, HF/6-31G**, HF/6-311G**. We have also performed a geometric optimization calculation at the B3LYP/6-31G, B3LYP/6-31G** and B3LYP/6-31G** level. The NMR isotropic shielding constants were calculated using the standard GIAO (Gauge-Independent Atomic Orbital) approach of Gaussian 03 program package [98].
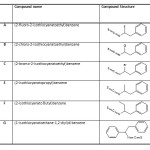 |
Table 1: Chemical shifts of the considered compounds were calculated at the same level using the Gauge- Included Atomic Orbital (GIAO) approach. Click here to View table |
Molecular mechanics (Monte Carlo simulation)
The Metropolis implementation of the Monte Carlo algorithm has been developed by studying the equilibrium thermodynamics of many-body systems. Choosing small trial moves, the trajectories obtained applying this algorithm agree with those obtained by Langevin’s dynamics. This is understandable because the Monte Carlo simulations always detect the so-called “important phase space” regions which are of low energy. Because of imperfections of the force field, this lowest energy basin usually does not correspond to the native state in most cases, so the rank of native structure in those decoys produced by the force field itself is poor.
In density function theory the exact exchange (HF) for a single determination is replaced by a more general expression the exchange correlation functional, which can include terms accounting for both exchange energy and the electron correlation, which is omitted from Hartree-Fock theory:
![]()
where Eχ(p) is the exchange function and EC(p) is the correlation functional.
Langevin dynamics (LD) simulation
The Langevin equation is a stochastic differential equation in which two force terms have been added to Newton’s second law to approximate the effects of neglected degrees of freedom .These simulations can be much faster than molecular dynamics.The molecular dynamics method is useful for calculating the time dependent properties of an isolated molecule. However, more often, one is interested in the properties of a molecule that is interacting with other molecules.
Results and Discussion
We used different force field to determination of energy and other type of geometrical parameters, on the particular SWCNT, and DWCNTs Because of the differences among force fields, the energy of a molecule calculated using two different force fields will not be the same.In the process of investigating the combination of particular molecules and CNT to achieve more information about these important gene transfer systems, we attempted to construct different (2-fluoro-2- isothiocyanatoethyl) benzene, (2-chloro-2- isothiocyanatoethyl) benzene, (2-iso-thiocyanatopropyl) benzene, (2-bromo-2-isothiocyanatoethyl)benzene and (2-isothiocyanatoButyl)benzene and link them individually to outer surface of SWCNT to develop practical application of these molecules to interact with SWNTs and DWCNTs. These coupling have been performed between C atom of nanotubes and halogen of molecules.In Fig.1 the couplings between these molecules and single wall nano tubes with outer surface of SWNT has been displayed. Optimizations of these structures have been performed by using UFF and AM1[71].
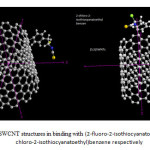 |
Figure 1: (5,5) and (9,9) SWCNT structures in binding with (2-fluoro-2-isothiocyanatoethyl)benzene and (2-chloro-2-isothiocyanatoethyl)benzene respectively Click here to View figure |
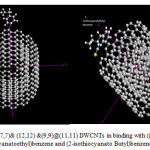 |
Figure 2: (7,7)& (12,12) &(9,9)@(11,11) DWCNTs in binding with ((2-bromo-2-isothiocyanatoethyl)benzene and (2-isothiocyanato Butyl)benzene respectively Click here to View figure |
So, it is not reasonable to compare the energy of one molecule calculated with a particular force field with the energy of another molecule calculated using a different force field. In this study difference in force field illustrated by comparing the energy of calculated by using force fields, MM+, Amber and OPLS. Theoretical energy values using different force fields, which is the combination of attraction van der Waals forces due to dipole-dipole interactions and empirical repulsive forces due to Pauli repulsion has been demonstrated in Table1.Since the solute dipole moment induces a dipole moment in opposite direction in the surrounding medium.
Table 1: Calculated dipole moment and optimized energy parameters(relative) with different levels of Theory, which are affected by different dielectric constants.
|
Media |
Temperature Dipole moment Energy |
|||||||||
|
a |
|
305 5.54 -51.1211 |
307 4.44 -51.0136 |
309 3.89 -51.083 |
311 7.9 -55.373 |
313 4.5 -63.534 |
315 3.22 -47.443 |
317 4.33 -56.610 |
319 4.25 -33.968 |
321 3.45 0.0 |
|
b |
1.02 -34.927 |
2.714 -35.496 |
2.25 -37.185 |
1.34 -49.365 |
1.276 -38.160 |
3.95 -30.994 |
1.916 0.0 |
1.653 -30.44 |
2.997 -35.442 |
|
|
c |
6.4 -36.34 |
5.03 -25.247 |
3.83 -35.165 |
3.55 -33.345 |
5.84 -34.55 |
4.33 0.0 |
2.44 -34.555 |
5.31 -17.361 |
6.66 -26.231 |
|
|
e |
1.152 -39.48 |
4.194 -42.35 |
3.261 -47.36 |
3.416 -55.33 |
2.44 0.0 |
3.565 -36.26 |
3.461 -37.26 |
2.44 -34.55 |
2.651 -38.98 |
|
|
f |
2.84 -31.72 |
1.68 -34.31 |
3.55 0.0 |
2.58 -23.82 |
1.27 -33.259 |
3.28 -43.156 |
2.77 -46.18 |
1.94 -40.46 |
2.961 -62.287 |
|
|
g |
3.218 -51.27 |
2.726 -38.561 |
3.943 -56.234 |
3.149 -54.48 |
3.242 -42.57 |
5.645 -35.545 |
3.345 -35.72 |
3.83 -43.635 |
4.46 0.0 |
|
|
|
|
HF |
B3LYP |
Molecular Mechanic Methods |
|
E |
|
|
||
|
(7,7)SWNTS |
|
E×10-5 Sto-3g 3-21g |
μ Sto-3g 3-21g |
E Sto-3g 3-21g |
μ Sto-3g 3-21g |
Dielectric constant |
MM+ |
Amber |
OPLS |
|
|
(11,11)SWNTS |
|
-35.24 |
29.85 |
-42.36 |
43.85 |
1 |
472.55 |
250.32 |
317.77 |
|
|
|
|
-35.49 |
75.59 |
-72.65 |
68.61 |
78.00 |
422.56 |
862.79 |
375.63 |
|
|
(7,7)@(12,12)DWNTS |
|
-52.55 |
31.51 |
-56.29 |
36.79 |
32.00 |
540.69 |
176.17 |
364.42 |
|
|
|
|
-42.36 |
47.55 |
-50.63 |
23.30 |
24.00 |
224.12 |
147.42 |
516.47 |
|
|
(5,5)@(10,10)DWNTs |
|
-34.57 |
34.69 |
-62.65 |
16.81 |
70.00 |
541.65 |
360.79 |
247.74 |
|
|
|
|
-22.76 |
64.14 |
-34.03 |
23.34 |
72.00 |
545.18 |
149.87 |
391.12 |
|
 |
Figure 2: The chart of optimized energy using theoretical methods in various media, for SWNTs and DWNTS Click here to View figure |
Also, polarization of the medium in turn polarizes the charge distribution in the solvent. The dipole moment value of SWCNT in different solvent media at same temperatures has been reported.
References
- Brzezinski, W.Medelewski, P. Z Pflanzenzuchtg.1984, 93, 177-183.
- Fahey, J. Zalcmann,A.T.Talalay, P. Phytochemistry.2001, 56, 5–51
- Nugon-Baudon, L.Rabot, S.Flinois, J-P.Lory, S.Beaune, P. Br. J. Nutr.1998,80, 231–234,
- Grubbs, C. J.Steele, V. E.Casebolt, T.Juliana, M.M.Eto, I.Whitaker,L.M.Dragnev,K. H.Kelloff, G. J.Lubet, R. L. Anticancer Res.1995,15,709– 716
- Nakagawa, H.Tsuta, K.Kiuchi, K.Senzaki, H.Tanaka, K.Hioki, K.Tsubura, A. Carcinogenesis (Lond.),2001,22,891–897
- Wattenberg, L. W. Cancer Res.1981,41, 2991–2994
- Bresnick, E.Birt, D. F.Wolterman, K.Wheeler, M.Markin, R. S. Carcinogenesis (Lond.),1990, 11,1159–1163
- Stoewsand, G. S. Anderson, J. L. and Munson, L. Cancer Lett.1988, 39,199–207
- Bogaards, J. J.Verhagen, H. Willems, M. van Poppel, G.van Bladeren, P. J Carcinogenesis (Lond.),1994, 15, 1073–1075
- M. Monajjemi & J. Najafpour , Fullerenes, Nanotubes, and Carbon Nanostructures, 2014,22(6),575–594,.
- Monajjemi, M.Baei, M.T.Mollaamin, F. Russian Journal of Inorganic Chemistry. 2008, 53 (9), 1430-1437
- Monajjemi, M. Rajaeian, E. Mollaamin, F.Naderi, F.Saki, S. Physics and Chemistry of Liquids. 2008, 46 (3), 299-306
- Monajjemi, M. Seyed Hosseini, M. Journal of Computational and Theoretical Nanoscience 2013 ,10 (10), 2473-2477
- Yahyaei ,H.Monajjemi, M. Fullerenes, Nanotubes, and Carbon Nanostructures.2014, 22(4), 346–361
- Monajjemi, M .Jafari Azan, M.Mollaamin, F. Fullerenes, Nanotubes, and Carbon Nanostructures.2013, 21(6), 503–515
- Bhupesh Bishnoi.Bahniman Ghosh ,Quantum Matter,2014, 3, 469-475
- SuleCelasun ,Rev. Theor. Sci. 2013,1, 319-343
- Nafisi, S.Monajemi, M.Ebrahimi, S. Journal of Molecular Structure.2004,705 (1-3) 35-39
- Monajjemi , M.Baheri ,H.Mollaamin ,F. Journal of Structural Chemistry.2011, 52(1), 54-59
- Monajjemi, M.Seyed Hosseini, M. Mollaamin, F. Fullerenes, Nanotubes, and Carbon Nanostructures. 2013, 21, 381–393
- DavideFiscaletti.AmritSorli ,Quantum Matter,2014, 3, 200-214
- Monajjemi, M. Boggs, J.E. J. Phys. Chem. A, 2013,117,1670 −1684
- Monajjemi , M. Honaparvar , B. KhaliliHadad ,B. Ilkhani ,AR. Mollaamin, F. Afr. J. Pharm. Pharmacol .2010, 4 (8), 521-529
- Piglosiewicz,B.;Vogelsang,J.Schmidt,S.JaePark,D.Groß,P.Lienau, C ,Quantum Matter.2014, 3, 297-306
- Monajjemi, M. Chemical Physics. 2013, 425, 29-45
- Fazaeli ,R.Monajjemi ,M.Ataherian ,F.Zare, K. Journal of Molecular Structure THEOCHEM.2002, 581 (1), 51-58
- Monajjemi, M.Mollaamin, F, J Clust Sci,2011,22,673.
- Anurag Srivastava, NileshiSaraf, and A. K. Nagawat , Quantum Matter.2013, 2, 401-407
- IijimaSumio. Nature (London), 1991,354, 56.
- S. Iijima, Nature, 1991,354, 56.
- S,Iijima,S.Ichihasi,T,Nature,1993,363, 603
- Bethune,D. S.Kiang,C. H.deVries,M. S. Gorman,G.;Vazques, J. Beyers,R,Nature ,1993,363, 605
- Ramanathan,T.Fisher,F.T.Ruoff,R.S.Brinson,L.C.Chem mater,2005 17, 1290
- Monajjemi, M .Sobhanmanesh, A .Mollaamin, F.Fullerenes, Nanotubes, and Carbon Nanostructures,2013, 21,47–63
- Monajjemi ,M.Karachi ,N.Mollaamin, F. Fullerenes, Nanotubes, and Carbon Nanostructures, 2014, 22,643–662
- Davide Fiscaletti,Rev. Theor. Sci.2013,1, 103-144
- Monajjemi, M. Mahdavian, L. Mollaamin, F. Bull .Chem.Soc.Ethiop ,2008, 22(2),1-10.
- Guo,J.-Y.. Xu,C.-X. . Sheng,F.-Y. .Jin, Z.-L.Shi,Z.-L. Dai,J. . Li,Z.-H. Quantum Matter,2013,2, 181-186
- Mollaamin ,F. Baei, MT. Monajjemi, M.Zhiani , R.;Honarparvar , B. Russian Journal of Physical Chemistry A, Focus on Chemistry,2008, 82, (13), 2354-2361
- Monajjemi, M.Ghiasi, R. Ketabi, S. Journal of Chemical Research.2004, 1,11-18
- Mahdavian,L. Monajjemi, M.Mangkorntong ,N.Fullerenes, Nanotubes and Carbon Nanostructures,2009, 17 (5), 484-495
- Mollaamin, F. Gharibe, S. Monajjemi, M. Int. J. Phy. Sci ,2011,6, 1496-1500
- Monajjemi, M . Faham, R. Mollaamin, F.Fullerenes, Nanotubes, and Carbon Nanostructures 2012 20, 163–169
- Monajjemi, M. Khaleghian, M. Tadayonpour, N. Mollaamin, F. International Journal of Nanoscience,2010, 9 (05), 517-529
- Mollaamin , F .Najafi ,F. Khaleghian, M. KhaliliHadad, B. Monajjemi ,M. Fullerenes, Nanotubes, and Carbon Nanostructures,2011,19, 653–667
- Monajjemi, M. Chegini , H. Mollaamin , F. Farahani ,P,Fullerenes, Nanotubes, and Carbon Nanostructures.2011,19, 469–482
- Monajjemi, M.Yamola ,H. Mollaamin,F.Fullerenes, Nanotubes, and Carbon Nanostructures 2014, 22, 595–603
- Monajjemi, M. Heshmata, M. Haeri, HH , Biochemistry (Moscow),2006, 71 (1), S113-S122
- Monajjemi, M. TheorChemAcc ,2015, 134:77 DOI 10.1007/s00214-015-1668-9
- A. Rubio. J.L. Corkill.M.L. Cohen. Phys. Rev. B ,1994, 49, 5081
- X. Blasé. A. Rubio.S.G. Louie. M.L. Cohen. Europhys.Lett,1994.28, 335.
- N.G. Chopra. J. Luyken. K. Cherry. V.H. Crespi. M.L. Cohen,S.G. Louie. A. Zettl, Science.1995 , 269, 966
- N.G. Chopra.A. Zettl.Solid State Commun.1998 ,105, 297
- J. Cumings. A. Zettl, Solid State Commun.2004, 129, 661
- R. Ma.Y. Bando. H. Zhu.T. Sato, C. Xu, D. Wu, J. Am.Chem. Soc.2002, 124, 7672,
- P.W. Fowler, K.M. Rogers, G. Seifert, M. Terrones, and H. Terrones, Chem. Phys. Lett. 1999, 299, 359
- Monajjemi, M . Falahati, M. Mollaamin, F. Ionics, 2013 , 19, 155–164
- Monajjemi , M. Heshmat ,M. Aghaei , H. Ahmadi , R. Zare,K. Bulletin of the Chemical Society of Ethiopia, 2007,21 (1)
- Monajjemi , M. Lee, V.S. Khaleghian, M. B. Honarparvar, B. F. Mollaamin, F, J. Phys.Chem. C. 2010, 114 (2010) 15315
- A. Rubio, J.L. Corkill, M.L. Cohen, Phys. Rev. B.1994, 49,5081
- X. Blase, A. Rubio, S.G. Louie, M.L. Cohen, Europhys. Lett.1994, 28, 335.
- N.G. Chopra, J. Luyken, K. Cherry, V.H. Crespi, M.L. Cohen,S.G. Louie, A. Zettl, Science ,1995, 269, 966.
- O.R. Lourie, C.R. Jones, B.M. Bartlett, P.C. Gibbons, R.S. Ruoff,W.E. Buhro, Chem. Mater.2000, 12 ,1808
- R. Ma, Y. Bando, T.Sato, Chem. Phys. Lett.2001,337 ,61.
- W. Han, Y. Bando.; K. Kurashima.; T. Sato, Appl. Phys. Lett.1998,73, 3085
- (a) D. Golberg. Y. Bando.M. Eremets. K. Takemura. K. Kurashima.H.Yusa, Appl. Phys. Lett.1996 ,69, 2045
- D.P. Yu, X.S. Sun, C.S. Lee, I. Bello, S.T. Lee, H.D. Gu, K.M. Leung, G.W. Zhou, Z.F. Dong. Z. Zhang. Appl. Phys. Lett.1998,72 , 1966.
- F.Jensen. H.Toftlund, Chem. Phys. Lett.1993, 201,89 , 94
- Monajjemi, M . Aghaie , H. Naderi , F. Biochemistry (Moscow).2007,72 (6), 652-657
- Monajjemi, M. Journal of Molecular Modeling , 2014, 20, 2507
- Davide Fiscaletti,Rev. Theor. Sci.2013 1, 103-144
- Monajjemi , M. Chahkandi ,B. Zare,K. Amiri, A. Biochemistry (Moscow),200,70, (3), 366-376
- Monajjemi, M . Afsharnezhad ,S. Jaafari , M.R. Abdolahi ,T. Nikosade ,A. Monajemi ,H.Russian Journal of physical chemistry A, 2007,2,1956-1963
- Monajjemi, M. Khaleghian, M, Journal of Cluster Science. 2011, 22(4), 673-692
- Mollaamin , F. Monajjemi , M , Journal of Computational and Theoretical Nanoscience. 2012, 9 (4) 597-601
- Monajjemi, M. Struct. Chem, 2012, 23 551.
- Jon M. Matxain. Jesus M. Ugalde.; M. D. Towler. and R. J. Needs. J. Phys. Chem. A 2003, 107, 10004-10010
- WU Haishun.XUXiaohong, JIAO Haijun, ZhangFuqiang .; JIA Jianfeng. Chinese Science Bulletin.2003, 48, 11 1102 1107
- Monajjemi, M . Ketabi ,S.Amiri, A. Russian Journal of Physical Chemistry , 2006, 80 (1), S55-S62
- M. Monajjemi . Robert Wayne Jr, J.E. Boggs, Chemical. Physics. 2014, 433,1-11
- Jon M. Matxain. Jesus M. Ugalde. M. D. Towler.and R. J. Needs. J. Phys. Chem. A 2003, 107, 10004-10010
- WU Haishun. XU iaohong. JIAO Haijun, ZHANG Fuqiang . JIA Jianfeng Chinese Science Bulletin.2003,48 (11), 1102 1107
- Monajjemi , M. Honarparvar, B. Monajemi, H.Journal of the Mexican Chemical Society, 2006, 50 (4), 143-148
- Monajjemi ,M. Mollaamin ,F. Journal of Computational and Theoretical Nanoscience,2012 ,9 (12) 2208-2214
- Monajjemi, M.Mahdavian, L. Mollaamin, F. Honarparvar, B. Fullerenes, Nanotubes and Carbon Nanostructures, 2010,18, 45–55
- Friedrich, B. J.D. Weinstein. R. Decarvalho . J.M. Doyle. Trap. J. Chem. Phys. 1999,110,2376-2383.
- Ramsay, N.E. Magnetic Shilding of Nuclei. J. Phys. Rev.1950, 78, 699-703.
- Frischend, M.J. J.B. Foresman,. Gaussian 94 user’ reference (Gaussian, Inc., Pittsburgh). 1995
- Ghalandari, B. Monajjemi, M. Mollaamin, F. Journal of Computational and Theoretical Nanoscience, 2011,8, 1212–1219
- Monajjemi , M.Khosravi , M. Honarparvar, B. Mollaamin, F. International Journal of Quantum Chemistry, 2011, 111, 2771–2777
- Monajjemi, M. Rajaeian, E. Mollaamin, F. Physics and Chemistry of Liquids,2008, 46, 299.
- Tahan, A .Monajjemi, M. Acta Biotheor,2011, 59, 291–312
- Monajjemi, M. Farahani, N. Mollaamin, F. Physics and Chemistry of Liquids 2012, 50(2) 161–172
- Monajjemi, M.Razavian, M.H. Mollaamin,F. Naderi,F. Honarparvar,B. Russian Journal of Physical Chemistry A , 2008 , 82 (13), 2277-2285
- Mollaamin , F. Varmaghani , Z. Monajjemi , M, Physics and Chemistry of Liquids. 2011,49, 318
- Monajjemi, M. Honarparvar, B. H. Haeri, H. Heshmat, M. Russian Journal of Physical Chemistry C, 2008, 80(1),S40-S44.
- Cheeseman, J.R. M.J. Frisch. F.J. Devlin .P.J.Stephens, Chemical Physics Letters, 1996,252 (3-4), 211-220.
- Naghsh,F, oriental journal of chemistry, 2015, 31(1)
- Chitsazan, A,oriental journal of chemistry, 2015, 31(1)
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery, J.A., Vreven, T., Kudin, K.N., Burant, J.C., Millam, J.M., Iyengar, S.S., Tomasi, J., Barone, V., Mennucci, B., Cossi, M., Scalmani, G., Rega, N., Petersson, G.A., Nakatsuji, H., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Klene, M., Li. X., Knox, J.E., Hratchian, H.P., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi. R., Pomelli, C., Ochterski, J.W., Ayala, P.Y., Morokuma, K., Voth, G.A., Salvador, P., Dannenberg, J.J., Zakrzewski, V.G., Dapprich, S., Daniels, A.D., Strain, M.C., Farkas, O., Malick, D.K., Rabuck, A.D., Raghavachari, K., Foresman, J.B., Ortiz, J.V., Cui, Q., Baboul, A.G., Clifford, S., Cioslowski, J., Stefanov, B.B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I.,Martin, R.L., Fox, D.J., Keith, T., Al-Laham, M.A., Peng, C.Y., Nanayakkara, A., Challacombe, M., Gill, P.M.W., Johnson, B., Chen, W., Wong, M.W., Gonzalez, C., and Pople, J.A., Gaussian 03, Revision B.03, Gaussian, Inc, Wallingford CT (2004).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


