Adsorption behavior of CO on pristine and doped B12P12 nanocage : a DFT study
Leila Hojatkashani
Department of Chemistry, College of Basic Sciences, Yadegar-e-Imam Khomeini (RAH) Branch, Islamic Azad University. Tehran. Iran
DOI : http://dx.doi.org/10.13005/ojc/310429
Article Received on :
Article Accepted on :
Article Published : 10 Dec 2015
Adsorption of the pollutant gas CO on B12P12 nanocage surface is studied through density functional calculations. HOMO and LUMO energy levels ,binding energies and energy bond gaps of three possible configurations of CO on pristine B12P12 has been calculated by means of B3LYP and M062X functional with 6-31g+ basis set.The results showed that there is no or very slight adsorption of CO molecule by pristine B12P12.To overcome the fault, CO adsorption is investigated on Al and N doped B12P12 nanocage with the same method and basis set.The electronic and structural parameters like HOMO and LUMO energy levels and binding energies of possible configurations are calculated and showed that doped B12P12 with both Al and N atoms have increased about % 1.7 with CO molecule which also indicates more Vander Waals attraction between CO and Al and N doped B12P12 nanocage.
KEYWORDS:Adsorption; CO on pristine
Download this article as:| Copy the following to cite this article: Hojatkashani L. Adsorption behavior of CO on pristine and doped B12P12 nanocage : a DFT study. Orient J Chem 2015;31(4). |
| Copy the following to cite this URL: Hojatkashani L. Adsorption behavior of CO on pristine and doped B12P12 nanocage : a DFT study. Orient J Chem 2015;31(4). Available from: http://www.orientjchem.org/?p=13245 |
Introduction
Detection and control of gases are important issues in environmental studies and much experiments have been made on the development of materials which can be used as gas-sensitive and dangerous chemical removal devices [1,2]. Carbon monoxide (CO) is one of the gases which has been vastly studied because it is one of the most important pollutant gases in the environment. The main sources of this gas are automobiles and industrial process. CO can effect dangerously on the human body and the ecosystem of animals. In most studies CO gas has been detected in a range of 4–10 ppm. This gas has no color and no odor [3].
Carbon nanotubes as known as CNTs can adsorb gases such as CO and NO molecules but the energy for adsorption of these two gases by CNTs are very weak [4,5]. Some quasi-one-dimensional nanotubes and III–V compound semiconductors which are isoelectronic to CNTs such as BN, AlN, BP and AlP have been investigated [6–10]. These materials may be good substitutes for detecting pollutant gases such as CO and NO. Theoretical studies of several (XY)n cages (X=B, Al, …and Y=N, P, …) showed the fullerene-like cages X12Y12 can be the most stable ones which demonstrate specific stability when n is 12 [11-12]. At first, BP nanocages with 4- and 6-membered rings have been synthesized, but later nanocages of B12N12 were synthesized by Oku et al. indicated that B12N12 nanocages have 4- and 6-membered rings of BN [13]. BP are polar materials because of the slight positive charges of boron (B) atoms and the slight negative charges of phosphorus (P) atoms. The BP is a semiconductor compound which can be used in high temperatures and hard conditions in optoelectronic and microelectronic devices because of their special properties and abilities [14].
In the first step of this work, in we investigate the interaction and the adsorption energy of CO molecule on pristine B12P12 nanocage using two density functional theory (DFT) calculations. As the next step, we continue calculating the interaction of Al and N doped B12P12 nanocage with CO molecule using same (DFT) calculations. Our porpose of this experiment is to examine the capability of B12P12 nanocage and its doped variations for detecting CO molecule and the effects of Al and N atoms on B12P12/CO on electronic level.
Computional Methods
By using DFT functional methods (B3LYP,M062x) with 6–31g* basis set, Geometry optimizations , energy calculations , and density of states (DOS) analysis of B12P12 , CO , three different CO/B12P12 complexes configurations , three doped B12P12 nanocages and their complexes with CO were performed in GAMESS suite of the program [15,18]. Vibration frequencies were also calculated at the same level to confirm that all the stationary points correspond to true minima on the potential energy surface .In this study, the dopant atoms are Aluminum and Nitrogen which are chosen from the third and fifth groups of the periodic table of elements, respectively. B3LYP is a basis set which is mostly used for investigations and calculations of nano structure topics [19–24] but in this paper M062x is used as well [16].
The adsorption energy (Ead) of CO is as follows:
Ead=Etot(CO/B12P12)- Etot(B12P12) – Etot(CO) ,
where E(CO/B12P12) is the total energy of a CO molecule adsorbed on the surface of B12P12 , and E(B12P12) and E(CO) are the total energies of B12P12 and the CO molecule , respectively .According to the definition, negative values of Ead correspond to exothermic adsorption process.
Results and Discussion
Adsorption of CO on pristine B12P12
Formation carbon monoxide from the free atoms requires separations and a charge transfer from the atom C to O, but before reaching the equilibrium bond distance, this charge transfer reverses. Politzer et al. calculated the MEP of the carbon monoxide and showed that negative regions belonged to both C and O atoms. It means both carbon and oxygen atoms have neucleophilic characters. [25]. Azizi et al. showed that the adsorption of CO molecule on CNTs via the oxygen head is stronger than that via its carbon head [26]. Figure 1a shows the MEP of the carbon monoxide.
In order to find the most stable CO-adsorbed configuration, several configurations were used for optimization including C head or O head located on top and parallel on B12P12 nanocage. In this report, CO molecule is optimized by using two DFT functional methods (B3LYP,M062x). the results can be seen in table 1.
The MEP of B12P12 is shown in figure 1b, it shows charge integration on top of P atoms on B12P12 surface. By comparing figures 1(a) and 1(b), it can be concluded that the most stable configuration of 0O and B12P12 is due O atom of CO molecule and B atom of B12P12 nanocage.
Figures 2 and 3 show the optimized structure of B12P12 nanocage by using B3LYP and M062x and also theirs DOS plot. According to these results, by using B3LYP and M062x methods, the optimized nanocage has HOMO–LUMO energy gap (Eg) of 3.70 eV and 5.54 eV, respectively (table2). It can be concluded that, with M062X as DFT functional HOMO–LUMO energy gap increased. The figures also show slight changes of angles in optimized B12P12 by changing from B3LYP to M062X.
Figure 4 shows three possible configuration of CO/B12P12 complexes. In theses configurations, CO molecule was located in 1.9 Å distance from B12P12 nanocage .In the first step of this work, , these three configurations are optimized by using DFT functional method B3LYP. Then their DOS plots and HOMO–LUMO energy gap (Eg) are determined and compared. The optimized complexes and their DOS plots are shown in figure 5.
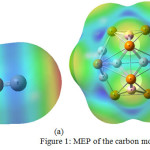 |
Figure 1: MEP of the carbon monoxide |
Table 1: total energy and optimazation method of CO
| Structure Method E total (a.u) |
| CO B3LYP -113.31 |
| CO M062X -113.27 |
Table 2: total energy, energy of HOMO and LUMO orbitals, Eg value of B12P12
| Structure Method E total (a.u) E HOMO(eV) E LUMO(eV) E g |
| B12P12 B3LYP -4394.65 -6.83 -3.13 3.70 |
|
B12P12 M062X -4394.14 -7. 98 -2.44 5.54
|
As it is shown in figure 5, in the three optimized configurations the distance between CO molecule and B12P12 nanocage are more than 1.9 Å . In the configuration (I), the distance between O atom of CO and B atom of B12P12 is 3.38Å. While in the configurations (II) and (III) the distance between C atom of CO and P atom of B12P12 is 3.81 Å and 3.77 Å ,respectively.
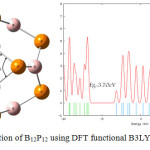 |
Figure 2: optimization of B12P12 using DFT functional B3LYP and its DOS plot (angles are in degree) Click here to View figure |
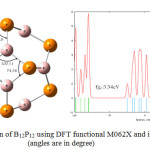 |
Figure 3: optimization of B12P12 using DFT functional M062X and its DOS plot (angles are in degree) Click here to View figure |
Comparing the DOS plots of configurations (I), (II) and (III), shows that the HOMO–LUMO energy gaps of the three configurations are the same as HOMO–LUMO energy gap of B12P12 which means inthose configurations no adsorption happened.
The optimization of B12P12 and the three CO/B12P12 complexes are repeated by using DFT functional method M062X. the Dos plot of pristine B12P12 showed increase of HOMO–LUMO energy gap from 3.70eV to 5.54eV (figures 2,3).
Figure 6 shows DOS plots and HOMO–LUMO energy gap (Eg) of the three configurations of CO/B12P12 complexes using M062X. The figure shows that HOMO–LUMO energy gaps in those complexes increased. But Again the HOMO–LUMO energy gaps of configurations (I), (II) and (III) are the same as HOMO–LUMO energy gap of pristine B12P12 which means inthose configurations there are no adsorption. These results show there is a same patern in adsortion between B12P12 and CO using two different DFT functional methods. Table 3 Shows all the results and comparisions
Also figure 6 shows in the configuration (I), the distance between O atom of CO and B atom of B12P12 is 3.21 Å. While in the configurations (II) and (III), the distance between C atom of CO and P atom of B12P12 are 4.02 Å 3.48 Å, respectively.
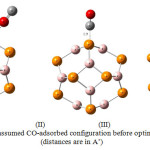 |
Figure 4: Assumed CO-adsorbed configuration before optimization(distances are in A˚) Click here to View figure |
Table 3: Total energy, binding energy, energy of HOMO and LUMO orbitals, Eg value, %Eg of B12P12 / CO configurations
| Structure Method E total E binding E HOMO E LUMO E g %E g (a.u) (Kcal/mol) (eV) (eV) |
| Config1 B3LYP -4507.96 0.00 -6.81 -3.11 3.70 0.00Config1 M062X -4507.41 0.00 -7.98 -2.44 5.54 0.00
|
| Config2 B3LYP -4507.96 0.00 -6.83 -3.13 3.70 0.00Config2 M062X -4507.41 0.00 -7.96 -2.42 5.54 0.00
|
| Config3 B3LYP -4507.96 0.00 -6.83 -3.13 3.70 0.00Config3 M062X -4507.41 0.00 -7.95 -2.41 5.54 0.00
|
Figures 5 and 6 show that by using M062X, the calculated Van der Waals bonds between B12P12 and CO in configurations (I) and (III) are slightly shorter than the calculated bonds using B3LYP. While the calculated bond in configuration (II) using M062X is longer than the calculated Van der Waals bond using B3LYP.
The effect of Al and N doping on B12P12 nanocage and their adsorption
In this level, we decided to study the effect of structure variation on the adsorption of B12P12 in comparison with its pristine form. To do such a doping, first one of the Boron atoms is replaced with an Aluminum atom which has more metallic characteristic. Second, one of the Phosphorous atoms is replaced with one Nitrogen atom and in third step, one of the Phosphorous atoms and one of the Boron atoms of pristine B12P12 are replaced with one Nitrogen and one Aluminum atom, respectively. In this step all optimizations are done using M062X method and 6-31g+ basis set.
At the first step, the effect of Al doping is investigated by the obtained results using M062X method and 6-31g+ basis set, the distance between O of CO molecule and Al atom in B12P12 is 2.51 Å which is shorter than the distance between B and O in the configuration (I) of CO/B12P12 using same method and basic set. In the next step, the interaction of N doping in B12P12 and CO molecule is calculated using same method and basic set. The results show that the distance between C of CO molecule and N atom in B12P12 is 3.43 Å which is shorter than the bond length between C atom and P atom in the configuration (II) of CO/B12P12 using same method and basic set. In the third step, the effect of Al and N doping together in pristine B12P12 on the adsorption of CO is being studied. As the figure shows after optimization the CO molecule is placed with O head on top of Al atom of the nanocage. The distance between O atom of CO and Al in the complex is 2.44 Å which is shorter than the bond length in the two previous configurations and between B atom and O atom in the configuration (I) of CO/B12P12 using same method and basic set. It can be cocluded that Al and N doped B12P12 nanocage has more interaction with CO molecule than other studied configurations in this paper.
Figures 7,8 and 9 show the change of angles in pristine B12P12 and its Al and N doped configurations. Slightly differences in angles of B12P12 and AlB11P12 can be seen but this differences in angles are increased in B12P11N and AlB11P11N. It can be the result of the effect of lone pair of doped N atom.
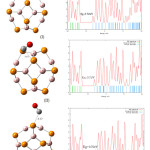 |
Figure 5: CO/B12P12 configurations after optimazation and their DOS plots (distances are in A˚) Click here to View figure |
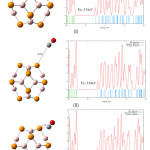 |
Figure 6: CO/B12P12 complex configuration1 (M062X) and its Dos plot (distances are in A˚) Click here to View figure |
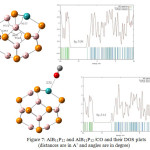 |
Figure 7: AlB11P12 and AlB11P12 /CO and their DOS plots (distances are in A˚ and angles are in degree)
|
The DOS plots of the Al- and N-doped B12P12 and their complexes with CO are shown in figures 7,8 and 9. As it is shown in those figures, the HOMO–LUMO energy gap (Eg) of AlB11P12, B12P11N and AlB11P11N after optimization are 5.06eV, 5.50eV and 5.27ev, respectively. It indicates no considerable change in the electronic properties of the three nanocages comparing with pristine B12P12. The results in these figures also show that interactions of the three doped nano cages with CO molecule. According to comparison Eg value of AlB11P12 with AlB11P12/CO , the calculated %Eg is 1.38% . It can conclude that there is some weak Van der Waals interactions in the system. Also comparing HOMO–Lomo energy gap of B12P11N with B12P11N /CO show us no difference in Eg value of the two structure so the calculated band energy gap variation is 0%. It shows no significant interaction exists between CO and N doped B12P12. Finally, According to the DOS plots and HOMO–LUMO energy gaps of AlB11P11N with AlB11P11N /CO ,a band energy gap variation of 1.7% is calculated .The result again shows a weak Van der Waals interactions exists in the system.
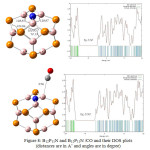 |
Figure 8: B12P11N and B12P11N /CO and their DOS plots (distances are in A˚ and angles are in degree) Click here to View figure |
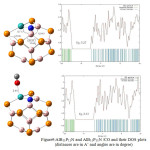 |
Figure 9:AlB11P11N and AlB11P11N /CO and their DOS plots(distances are in A˚ and angles are in degree) |
Table 4: total energy, binding energy, energy of HOMO and LUMO orbitals, Eg value and %Eg of Al and N doping B12P12 nanocages and their complexes with CO molecule
|
Structure Method E total E binding E HOMO E LUMO E g %E g (a.u) (Kcal/mol) (eV) (eV) |
| AlB11P12 M062X -4611.75 — -7.58 -2.52 5.06 —AlB11P11/CO M062X -4725.02 0.00 -7.45 -2.32 5.13 %1.38 |
| B12P11N M062X – 4107.63 — -7.73 -2.23 5.50 —B12P11N/CO M062X -4220.90 0.00 -7.69 -2.19 5.50 %0
|
| AlB11P11N M062X -4325.23 — -7.58 -2.30 5.28 —AlB11P11N/CO M062X -4438.51 -6.27 -7.46 -2.09 5.37 %1.70 |
Conclusion
The behavior of CO molecule adsorbed on the external surface of B12P12 nanocage cluster was studied using two DFT functional methods, B3LYP and M062X. Calculating the HOMO/LUMO energy gap and adsorption energy of B12P12 and its three configuration with CO with two DFT functional methods showed no significant change in the HOMO-LUMO gap of the nanocage .Again the behavior of CO molecule was detected on Al and N doped B12P12 nanocage. The results showed that adsorption of CO on Al and N doped B12P12, above the Aluminum atom of the surface as AlB11P11N/CO has the most stable state of adsorption and most increased in the HOMO-LUMO gap of the other Al and N doped B12P12 configurations. The %Eg of AlB11P11N/CO is calculated %1.7 which less than %7-8. As the result, B12P12 cannot be a potential efficient gas sensor for CO detection.
References
- Chang, H; Lee, J.D.; Lee, S.M.; Lee, Y.H. Appl. Phys. Lett. 2001.79. 3863-3865.
- Lu, J.; Nagase, S.; Maeda, Y.; Wakahara, T.; Nakahodo, T.; Akasaka, T.; Yu, D.; Gao, Z.; Han, R.; Ye, H. Chem. Phys. Lett. 2005 .405 . 90-92.
- Xie, Y.; Huo, Y. P.; Zhang, J. M. Applied Surface Science. 2012 .258 .6391–6397
- Zurek, E.; Autschbach, J. J. Am. Chem. Soc. 2004.126 .13079-13088.
- Nojeh, A.; Lakatos, G.W.; Peng, S.; Cho, K.; Pease, R.F.W. Nano Lett. 2003.3. 1187-1190.
- Su, C.Y.; Chu, W.Y.; Juang, Z.Y.; Chen, K.F.; Cheng, B.M.; Chen, F.R.; Leou, K.C.; Tsai, C.H. J. Phys. Chem. C . 2009.113. 14732-14738.
- Zhao, M.; Xia, Y.; Tan, Z.; Liu, X.; Li, F.; Huang, B.; Ji, Y.; Zhang, D.; Mei, L. Chem. Phys.Lett. 2004.389 . 160-164.
- Qian, Z.; Hou, S.; Zhang, J.; Li, R.; Shen, Z.; Zhao, X.; Xue, Z.; Physica E. 2005. 30. 81-85.
- Ahmadi, A.; Beheshtian, J.; Hadipour, N.L. Physica E. 2011.43. 1717-1719.
- Mirzaei, M.; Mirzaei, M. J. Mol. Struct. 2010. 951. 69-71.
- Strout, D.L. J. Phys. Chem. A. 2000. 104. 3364–3366
- Wang, R.; Zhang, D.; Liu, C. Chem. Phy.s Lett. 2005. 411.333–338.
- Oku, T.; Nishiwaki, A.; Narita, I.Sci. Technol. Adv. Mater .2004.5.635–638
- Ferreira, V.; Alves, H. J. Crys. Grow. 2008. 310. 3973–3978
- Zhao, Y.; Truhlar, D.G., Theor. Chem. Account. 2006. 120. 215–241.
- Wu, H.S.; Xu, X.H., Strout, D.L.; Jiao, H. J. Mol. Model .2005.12:1-8.
- DeMenezes, V.M.; Fagan, S.B.; Zanella, I.; Mota, R. Microelectron J. 2009.40. 877–879.
- Zhao, Y.; Truhlar, D.G. J. Chem. Theor. Comput .2008. 4.1849–1868
- Ni, M.Y.; Zeng, Z.; Ju, X. Microelectron J. 2009. 40. 863–866
- Ahmadi, A.; Beheshtian, J.; Hadipour, N.L. Physica E. 2011. 43.1717–1719.
- Ahmadi, A.; Beheshtian, J.; Hadipour, N.L. Struct. Chem. 2011. 22. 183–188.
- Tabtimsai, C.; Keawwangchai, S.; Wanno, B.; Ruangpornvisuti, V. J. Mol. Model . 2012. 18. 351-358.
- Beheshtian, J.; Soleymanabadi, H.; Kamfiroozi, M.; Ahmadi, A. J. Mol. Model. 2012. 18. 2343-2348.
- Oubal, M.; Picaud, S.; Rayez, M.; Rayez; J. Surf. Sci. 2010 . 604 . 1666-1673.
- Politzer, P.; Kammeyer, C.W.; Bauer, J.; Hedges, W.L. J. Phys. Chem. 1981.85 . 4057-4060.
- Azizi, K.; Hashemianzadeh, S.M.; Bahramifar, S. Curr. Appl. Phys. 2011.11 .776-782.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


