Mechanism of the Polymerization of Styrene and 2.3 Dihydro-4H-Pyran Catalyzed by H3PW12O40 Catalyst.
Ahmed Aouissi*, Zeid Abdullah Al-Othman and Hassan Al-Shehri
Chemistry Department, College of Science, King Saud University. Riyadh. KSA. Correspondence E-mail: aouissed@yahoo.fr
DOI : http://dx.doi.org/10.13005/ojc/310349
Article Received on :
Article Accepted on :
Article Published : 07 Aug 2015
The bulk copolymerization of styrene (ST) and 2.3 Dihydro-4H-pyran (DHP) catalyzed by 12-tungstophosphoric (H3PW12O40) acid was investigated. The polymerization was achieved in mild conditions by a very simple procedure and the catalyst was recovered by simple filtration. The obtained polymers were characterized by means of FTIR, 1H-NMR, viscosity measurement, differential scanning calorimetry (DSC), and thermogravimetry (TGA). The results indicated the formation of a copolymers and a homopolymer. The polymerization involved exclusively C=C bonds of the vinyl cyclic ether. Propagation by ring-opening was not observed. The effect of the temperature and the DHP proportion on the polymerization showed that low temperatures and high DHP proportion increased the polymerization conversion.
KEYWORDS:cationic polymerization; styrene; vinyl ethers; heteropoly acids
Download this article as:| Copy the following to cite this article: Aouissi A, Al-Othman Z. A, Al-Shehri H. Mechanism of the Polymerization of Styrene and 2.3 Dihydro-4H-Pyran Catalyzed by H3PW12O40 Catalyst. Orient J Chem 2015;31(3). |
| Copy the following to cite this URL: Aouissi A, Al-Othman Z. A, Al-Shehri H. Mechanism of the Polymerization of Styrene and 2.3 Dihydro-4H-Pyran Catalyzed by H3PW12O40 Catalyst. Orient J Chem 2015;31(3). Available from: http://www.orientjchem.org/?p=10168 |
Introduction
Polystyrene derivatives that contain functional groups are of great interest owing to their excellent properties. That is why they have found a wide range of commercial applications [1- 3]. These kinds of copolymers can be obtained by copolymerization of styrene with polar monomers. Among the polar monomers used, cyclic ethers are of major interest in the field of polymer chemistry. In fact, due to their high polarizability and flexibility, polyethers constitute very important soft segments for producing thermoplastic elastomers such as polyesters and polyurethanes. They represent a key ingredient in the production of a variety of elastomeric products. Therefore, they have been the subject of a large number of papers [4-6]. Due to the polar nature of the ether groups in the polyether segments and their ability to form hydrogen bonds, these polyether segments may form intra- as well as inter-molecular hydrogen bonds. Copolymerization of styrene and cyclic ethers is an effective method for imparting new functional properties to existing polystyrene without altering their inherent properties. The polymerization could be initiated by electrophilic agents such as Brønsted acids (HCl, H2SO4, HClO4, etc.) and also by Lewis acids (AlCl3, BF3 OEt2, TiCl4, etc.). However, the protonic acid catalysts used are corrosive and not recoverable, and the Lewis acids required great amount. Increasing environmental concerns in recent years call for a more effective catalytic process. Solid Brønsted acids with superacidic character, such as the Keggin-type heteropolyacids and their acidic salts are known as highly active catalysts [7]. They catalyze various reactions much more effectively than the conventional protonic acids [8]. That is why heteropolyacids have been used as catalysts to induce the polymerization of various monomers such as cyclic ethers, styrene, acetals, urethanes and lactones [9-15]. In this research work we report the copolymerization of ST and DHP. The effect of the monomer ratio and reaction temperature on the copolymer composition was investigated.
Material and Methods
Catalyst preparation
12-Tungstophosphoric acid, H3PW12O40.13H2O (abbreviated H3PW12O40) was prepared according to a now well-known method [16]. Purity was checked by IR and thermogravimetry,
Polymerization procedure
The copolymerization of ST and DHP was carried out in a stirred flask fitted with a condenser. Typically, a fixed amount of catalyst was added to 10 mL of the DHP and ST monomer mixture under stirring at the desired temperature (30oC) until the viscosity prevented it from occurring. The resulting precipitated polymer mixture was then dissolved in chloroform. After removing the catalyst by filtration, the poly[(DHP)x-( ST)1-x] was precipitated by addition of methanol, filtrated off, dried at 40-50oC under vacuum, and weighed. Then after the filtrate was placed under vacuum to remove the choroform and the poly(DHP) was precipitated.
Characterization
The keggin structure of 12-tungstophosphoric acid H3PW12O40.13H2O was checked by infrared (IR). IR spectra were recorded with an infrared spectrometer GENESIS II- FTIR (4000-400 cm-1) as KBr pellets. The number of protons was checked by means of thermogravimetry (TGA). 1H NMR spectra was recorded on a Bruker Avance 400 MHz spectrophotometer using 5mm NMR tubes using deuterated acetone-D6 as solvent. The glass transition temperature of the synthesized polymers was measured with a differential scanning calorimeter (Shimadzu DSC 60) instrument, previously calibrated with indium. Samples weighing between 10 and 12 mg were packed in aluminum DSC pans before placing in the heating DSC cell. The samples were heated from -100 to 200oC at a heating rate of 10oC /min. All data were collected from the third scan run.
Molecular weight measurements were determined by means of a HT-GPC, Model 430, Viscotek, Houston, TX USA gel permeation chromatograph (GPC) with CLM6210 column.
Results and Discussion
Characterization of the catalyst
The IR spectra have been assigned according to Ref [17].The main characteristic features of the Keggin structure are observed at 1080-1060 cm-1 (νas P-Oa), at 990-960 cm-1 (νas W-Od), at 900-870 cm-1(νa W-Od-W), and at 810-760 cm-1(νas W-Oc-W). The thermogravimetric analysis showed the two types of water molecules observed on heteropolyacids: crystallization and constitutional water [18]. Loss of crystallization water (13 H2O) occurs before 200oC, whereas loss of constitutional water, resulting from the protons bounded to the polyanions external oxygens, occurs above 300oC.
Characterization of the synthesized polymers
The polymerization of the ST and the DHP has led to a mixture of polymers, the poly(DHP) partially soluble and the poly[(DHP)x-( ST)1-x] insoluble in methanol-chloroform mixture. The poly(DHP) was obtained with higher yield but with lower molecular weight and a narrow polydespesrity than the poly[(DHP)x-( ST)1-x] (Table 1). This result indicated that the homopolymerization of DHP monomer is more favorable than the ST monomer.
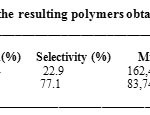 |
Table 1: Characteristics of the resulting polymers obtained over H3PW12O40 at 30oC. Click here to View table |
Fourier Transform Infrared Spectroscopy (FTIR)
The FTIR spectrum of poly[(DHP)x-( ST)1-x] is depicted in Figure 1. The band at 696 cm-1, 754 cm-1, and 3026 cm-1 were assigned to the C-H band of the cycle. The of the bands at 1452, 1492, 1600 cm-1 are assigned to the aromatic C=C. It is worth noting that the absence of the bands at 1224 cm-1 and 1647 cm-1 assigned to the double bond C=C in the 2,3 dihydro-4H-pyran (DHP) and the presence of the band at 1084 cm-1 assigned to the ether bond C-O-C indicate that the propagation process of the copolymerization by using this super acid exclusively involves the reaction of carbon–carbon double bonds. Copolymerization involving ring opening has not occurred. The FTIR spectrum of poly(DHP) polymer shows the presence of intense bands at 2846 and 2941 cm-1 which were assigned to the C-H band of the (CH2) of the DHP cycle. The presence of the intense band at 1087 cm-1, assigned to the ether bond C-O-C, and the absence of the bands at 1224 and 1647 cm-1, assigned to the double bond C=C, and indicates that the polymerization has occurred via the double bond but not by ring opening.
![Fig. 1: FTIR spectra of poly[(DHP)x-( ST)1-x] and poly(DHP) obtained at 30oC over H3PW12O40 catalyst.](http://www.orientjchem.org/wp-content/uploads/2015/08/Vol31_No3_Mech_Ahm_Fig1-150x150.jpg) |
Figure 1: FTIR spectra of poly[(DHP)x-( ST)1-x] and poly(DHP) obtained at 30oC over H3PW12O40 catalyst. Click here to View figure |
1H NMR
1H NMR of poly[(DHP)x-( ST)1-x]
The 1H-NMR spectrum (Figure 2) shows a wide band between 1.0 and 2.0 ppm corresponding to CH2 (b) and a wide band between 3.0 and 4.0 corresponding to the CH2 (a) neighbor of the oxygen atom in the heterocycle, and a wide band between 6.5 and 7.5 corresponding to the aromatic CH. This results indicate the formation of poly(St-DHP). It is worth noting that the absence of both the signal of the protons of the double bond and those of the aldehyde implies that the polymerization of the DHP monomer occurred by propagation via the double bonds but not via ring opening (Scheme 1). These results are in agreement with those of IR.
1H NMR of poly(DHP)
The 1H-NMR spectrum (Figure 3) shows a wide band between 1.0 and 2.0 ppm corresponding to CH2 (b) distant to the oxygen atom and a wide band between 3.0 and 4.0 corresponding to the CH2 (a) neighbor of the oxygen atom. This result indicates the formation of poly(DHP). In this case also, the signal corresponding to the protons of the double bond and that corresponding to the proton of the aldehyde were not observed in the spectrum indicating that the polymerization occurred by propagation via the double bonds and not via ring opening (Scheme 2).
![Fig. 2: 1H-NMR spectrum of Poly[(DHP)x-( ST)1-x] in CD3COCD3 .](http://www.orientjchem.org/wp-content/uploads/2015/08/Vol31_No3_Mech_Ahm_Fig2-150x150.jpg) |
Figure 2: 1H-NMR spectrum of Poly[(DHP)x-( ST)1-x] in CD3COCD3 . Click here to View figure |
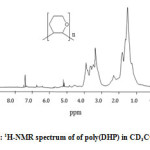 |
Figure 3: 1H-NMR spectrum of of poly(DHP) in CD3COCD3 Click here to View figure |
Scanning Calorimetry Analyses (DSC)
The DSC thermograms of poly[(DHP)x-( ST)1-x] and poly(DHP) are shown in Figure 4. It can be seen from the curves that the glass transition temperature value (Tg) is around 35oC for both polymers. The poly[(DHP)x(ST)1-x] has approximatively the same value of Tg with the poly(DHP) because the poly[(DHP)x(ST)1-x] is almost composed of DHP units, it contains only small units of styrene monomer.
![Fig. 4: DSC heating thermograms of poly[(DHP)x(ST)1-x] and poly(DHP) performed at heating rate of 10oC/min.](http://www.orientjchem.org/wp-content/uploads/2015/08/Vol31_No3_Mech_Ahm_Fig4-150x150.jpg) |
Figure 4: DSC heating thermograms of poly[(DHP)x(ST)1-x] and poly(DHP) performed at heating rate of 10oC/min. Click here to View figure |
Thermogravimetric analyses (TGA)
The TG curves for the poly[(DHP)x(ST)1-x] and the poly(DHP) are presented in Figure. 5. It can be seen from the figure that the decomposition of both polymers occurs at around 500oC. Although both polymers underwent similar stages of weight loss, the value of their weight loss are different. In fact, the poly[(DHP)x(ST)1-x] lost 90.9% of its initial weight, whereas the poly(DHP) lost 78.7%. The bigger weight loss of the poly[(DHP)x(ST)1-x] compared to the poly(DHP) might be due to the elimination of polystyrene oligomers present along with the poly(DHP).
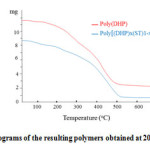 |
Figure 5: TGA thermograms of the resulting polymers obtained at 20ºC/min heating rate. Click here to View figure |
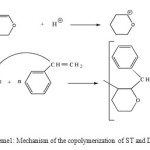 |
Scheme 1: Mechanism of the copolymerization of ST and DHP |
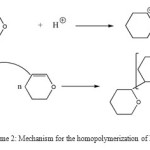 |
Scheme 2: Mechanism for the homopolymerization of DHP. Click here to View scheme |
Influence of DHP proportions on the polymerization conversion.
Figure 6 depicted the dependence of the conversion on the DHP mole fraction. It can be seen that the yield increased linearly with the proportion of DHP Monomer. This result indicated that the DHP is more reactive than the ST when the polymerization is catalyzed by brønsted acids. The high reactivity of the DHP monomer compared to that of the ST monomer can be explained by the fact that double bond of the heterocycle is activated by the oxygen in the vinyl ether cycle. That is why the polymerization led to the poly(DHP) homopolymer and to the poly[(DHP)x(ST)1-x] copolymer mainly composed of DHP units.
 |
Figure 6: The dependence of the copolymerization conversion on the DHP monomer mole fraction ratio. Click here to View figure |
Influence of temperature on the polymerization conversion.
Based on the fact that the reaction temperature can influence the reactivity of the monomers, we have investigated its effect on the conversion of the polymerization. The polymerization reactions were carried out in the temperatures ranging from 20–60 °C using an equimolar composition of monomers. The results illustrated in the Figure 7 indicated that the conversion decreased with the increase of the temperature. As for the polymer composition, it has been found that the yield of the poly(DHP) in the resulting polymers mixture was higher than that of the poly[(DHP)x(ST)1-x] (table 1). In addition, the proportion of DHP monomer was predominant in the poly[(DHP)x(ST)1-x]. This result is in agreement with that of Masuda et al [19]. In fact, in their study of the cationic copolymerizations of a pair of monomers having different structures such as vinyl ethers/styrene derivatives, the authors found that the propagating carbocation prefers to react with a monomer having a structure more similar to the carbocation. This result is also in agreement with that that of Yamamoto et al [20]. In the opinion of the authors, a carbocation produced from a vinyl ether reacts preferentially with a vinyl ether and a carbocation produced from a styrene derivative with a styrene derivative. The authors define this sort of phenomenon as “selectivity of carbocation” which leads to a difficulty of cross propagation and a formation of block-like copolymers in cationic copolymerizations.
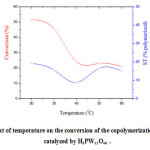 |
Figure 7: Effect of temperature on the conversion of the copolymerization. (Reaction catalyzed by H3PW12O40 . Click here to View figure |
Catalyst recycling
The continued pressure on petrochemical industries to eliminate or to reduce chemical waste in order to be more environmentally sensitive has motivated researchers to do much effort not only to develop performant catalysts but also to find ways to recover them. In fact, a recoverable catalyst will generate less waste and will be a key step toward the development of more efficient processes. In this context we have showed that H3PW12O40 can be recovered easily from the polymer products. The characterization by IR is depicted in the Figure 8. It can be seen from the figure that the spectrum of the used catalyst was not different of that of the fresh catalyst. Thus the Keggin structure of the used catalyst was intact. This result indicates that heteropolyacids can be used as green catalysts.
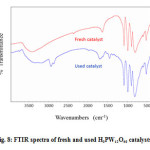 |
Figure 8: FTIR spectra of fresh and used H3PW12O40 catalysts. Click here to View figure |
Conclusion
The present work shows that H3PW12O40 is an effective solid acid catalyst for the cationic polymerization of Styrene and 2.3 Dihydro-4H-pyran. The polymerization of ST and DHP monomers led to a mixture of two easily separable polymers, the poly(DHP) soluble and the poly[(DHP)x(ST)1-x] insoluble in methanol/ chloroform mixture.
The conversion of the polymerization depends on the reaction temperature and on the DHP/ST ratio. High conversions were obtained at lower temperature and high DHP/ST ratio. DHP monomer was more reactive than the ST monomer.
The propagation process of the polymerization by using heteropolyacids exclusively involved the reaction of carbon–carbon double bonds. Polymerization involving ring opening was not observed.
The use of H3PW12O40 heteropolyacid as solid acid catalyst represents a more environmentally friendly alternative for the polymerization process. The polymerization proceeds in mild conditions by a very simple procedure, and the catalyst is recovered by simple filtration.
Acknoledgment.
The Authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding the work through the research group project No RGP-VPP-116.
References
- KE, D.M.; Zhang, L.; Wu, Q. Petrochemical Technology. 2003, 32(5), 438-434.
- Choudhary, M. S.; Varma, I. K. Macromol Sci Chem A. 1983, 20,771-779,
- Schoonbroad, H.A.S.; Aerdts A. M.; Germar A. L.; der Velden G.P.M. Macromolecules. 1995, 28, 5518-5525.
- Clark, J. S.; Elustondo, F.; Trevitt, G.P. ; Boyall, D. ; Robertson, J.; Blake, A.J.; Wilson, C.; Stammen, B. Tetrahedron. 2002, 58, 1973–1982.
- Jayakannan, M.; Ramakrishnan, S. Macromol. Rapid Commun. 2001, 22, 1463– 3. 1473.
- Chow, H.F.; Chan, I.; Mak, C.C.; Ng, M.K. Tetrahedron. 1996, 52, 4277–4290.
- Kozhevnikova, E.F.; Kozhevnikov, I.V. J. Catal. 2004, 224, 164–169.
- Kozhevnikov, I. V. Appl. Catal. A: Gen. 2003, 256, 3–18. 65.
- Chen, Y.; Zhang, G.L.; Zhang, H.Z. J. Appl. Polym. Sci. 2000, 77, 3239–3246.
- Wu, Q.; Li, L.; Yu, Y.; Tang, X. Colloid. Polym. Sci. 2008, 286, 761–767.
- Aouissi, A.; Al-Deyab, S. S.; Hassan Al-Shahri. Molecules. 2010, 15,1398-1407.
- Cheng, G.; Fan, X.; Pan, W.; Liu, Y. Journal of Polymer Research. 2010, 17(6), 4. 847-851
- Morinaga, H.; Nishioka, Y.; Hatayama, M.; Nakabayashi, H. Chemistry Letters. 5. 2013, 42 (7), 747-749
- Mahha, Y.; Atlamsani, A.; Blais, J.C.; Tessier, M.; Brégeault, J.M.; Salles, L. J. Mol. Catal. A: Chemical. 2005, 234, 63–73.
- Chen, Y.; Zhang, G.L.; Zhang, H.Z. J. Appl. Polym. Sci. 2001, 82, 269–275.
- Rocchiccioli-Deltcheff, C.; Fournier, M.; Franck, R.; Thouvenot R. Inorg. Chem. 7. 1983, 22, 207–216.
- Rocchiccioli-Deltcheff, C.; Fournier, M. J. Chem. Soc., Faraday Trans. 1991, 87, 8. 3913–3920.
- Fournier, M.; Feumi-Jantou, C.; Rabia, C.; Herve, G.; Launay, S. J. Mater. Chem. 9. 1992, 2, 971–978.
- Masuda, T.; Higashimura, T.; Okamura, S. Polym. J. 1970, 1, 19-26.
- Yamamoto, K.; Higashimura, T. Polymer, 1975, 16, 815- 818.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


