Synthesis and Characterization of 2, 3, 7, 8, 12, 13, 17, 18, Octabromo 5, 10, 15, 20 tetra phenyl porphyrin
Kalpana Raikwar
Department of chemistry, Govt.P.G.College,Guna(M.P.)473001.
Corresponding Author E-mail: kalpanaraikwar45@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/310276
Article Received on :
Article Accepted on :
Article Published : 02 Jun 2015
Chromium(III) porphyrin and its derivative have been synthesized by bromination and demetallation of copper tetra phenyl porphyrin.All the complexes were characterized on the basis of their elemental analysis, UV-vis spectra and magnetic moment properties.The magnetic moment suggested that the complex in the range 3.79 BM indicating an octahedral structure. The comparison of the UV- vis spectra of the free base show that substitution of Br atom at β-pyrrole position lead to greater shift than substitution of electronegative group in the phenyl ring.
KEYWORDS:Characterization; magnetic moment; Dimethyl formamide; octa bromo tetraphenyl porphinato chromium
Download this article as:| Copy the following to cite this article: Raikwar K. Synthesis and Characterization of 2, 3, 7, 8, 12, 13, 17, 18, Octabromo 5, 10, 15, 20 tetra phenyl porphyrin. Orient J Chem 2015;31(2). |
| Copy the following to cite this URL: Raikwar K. Synthesis and Characterization of 2, 3, 7, 8, 12, 13, 17, 18, Octabromo 5, 10, 15, 20 tetra phenyl porphyrin. Available from: http://www.orientjchem.org/?p=8993 |
Introduction
Metalloporphyrins are important model compound for understanding the biological function of heme proteins1. Recent works have shown that stability of porphyrin catalysts is greatly enhanced by substitution of β-pyrrole and ortho position of aryl groups on porphyrin ring with different electronegative groups2. Some sterically hinderd porphyrin complexes have been found to be efficient catalyst towards alkene epoxidation, hydroxylation3 etc. Chloro tetrakis(2, 6 dichlorophenyl) porphinato iron(III) has been frequently employed as monoxygenase catalyst because of resistance of porphyrin to oxidative degradation4. Similarly a series of tetrakis (penta fluorophenyl) iron(III) complexes have been proved as efficient catalyst for selective air oxidation of alkanes5.
Much has been said about the chemistry of porphyrin complexes and the effect of various changes in their structural features. The catalytic activity of these complexes is a topic of continued research. Hence some Cr(III) complexes of tetra phenyl porphyrins having either the β-pyrrole, position or orthophyenyl site or both position occupied by halogen atoms mainly chlorine and bromine were synthesized. The complexes have been characterized by UV-visible spectroscopy, magnetic properties, elemental analysis.
There have been much interest in the electronic structure of porphyrins and related compounds.This interest stems in part from their biological significance and catalytic properties6,7.The biologically important porphyrin derivative are all metal porphyrins have also received much attention in connection with their intrinsically interesting spectroscopic magnetic and electrochemical properties.The last decades have witnessed an explosion of experimental studies of metal porphyrins which have yielded very useful information about their electronic structure and optical spectra,but it has not always been possible to provide a well reasoned explanation of the result obtained8-11.
Metalloporphyrins is a natural choice for the optical detection of volatile organic compound because of their open co-ordination sites for axial ligation and their intense coloration12,13.Using metal contres that span a range of chemical “hardness” and ligand binding affinity,a wide range of volatile analytes might be differentiable. It has been found that porphyrin show significant solvato chromic effect,even with weakly interacting vapours14.
Experimental
The chemical components needed for the synthesis of porphyrin complexes were pyrrole, p-chloro-anil, acetylacetone, copper acetate pyridine, pottasium carbonate, sodium meta-bisulphite, anhy. sodiumsulphate, triethyl bromide, hydrochloric acid, perchloric acid, sulphuric acid, chromium trichloride, carbontetrachloride, chloroform,, dimethyl formamide, tetrahydrofuran etc.
Synthesis of Cr(Iii) Porphyrins
Synthesis of 2,3,7,8,12,13,17,18 octabromo 5,10,15,20 tetra phenyl porphyrin and its Cr(III) derivatives
The Cr(III) derivatives of octa bromotetra phenyl Porphyrin were synthesised by first synthesising the free base i.e. octabromotetra phenyl porphyrin [H2(OBTPP)]. The free base was then metallated with chromium, the overall procedure involved the following steps-
Preparation of Cu(TPP)15
Copper tetraphenyl porphyrin was synthesised by taking tetra phenyl porphyrin [H2(TPP)]16 (500mg) in chloroform(100ml). Copper(II) acetate(200mg) in glacial acetic acid(50ml) was added to the above solution and the mixture was refluxed for 2hrs. The contents were concentrated to a volume of about 50-60ml and cooled to room temperature which resulted in crude copper-tetraphenyl porphyrin Cu(TPP) (about 450mg). The crude product was purified by column chromatography using neutral alumina and chloroform as eluent. On elution the unreacted tetraphenyl porphyrin was eluted out first, followed by pure Cu(TPP). The chloroform fraction containing Cu(TPP) was concentrated to obtain pure crystals of Cu(TPP)[2]. The formation of Cu(TPP) was monitored by UV-visible spectroscopy which give peaks-around 580, 541 and 417nm respectively confirming the formation of Cu(TPP) (yield=400 mg).
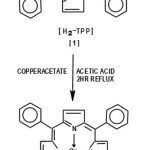 |
Figure 1 Click here to View figure |
Bromination of Copper tetraphenyl porphyrin Cu(TPP)17
The copper tetraphenyl porphyrins synthesised as above was converted into its octabromo derivative by the addition of liquid bromine (1.2ml)in chloroform(50ml) to a solution of Cu(TPP)(0.50 g) in chloroform:carbon tetrachloride (1:1 V/V) (500ml) in a conical flask. Bromine was added dropwise and slowly over a period of half hour, at room temperature. The contents were stirred for 4hours, followed by addition of pyridine 2.4ml in 40ml mixture of CHCl3:CCl4 in 1:1 ratio. The addition took about half hour and stirring continued for 12hours. The bromination process was monitored by UV-visible spectroscopy to ensure complete bromination.
The excess bromine was destroyed by addition of sodium metasulphite (200ml 20% aq. solution) to the system. The organic layer was separated using a separating funnel and the solution was dried over anhydrous sodium sulphate. The evaporation of solvent under reduced pressure resulted a green solid of copper octabromoteraphenyl porphyrin [Cu(OBTPP)]. The solid was dissolved in minimum amount of chloroform and column chromatography was done. The first fraction coming out of the column was collected. The removal of solvent yielded copper octabromotetraphenyl porphyrin(3) in purified form, yield (75%)
Demetallation of copper octabromo-porphyrin
Cu(OBTPP) synthesized above was demetallated to obtain a free base of octabromotetraphenyl porphyrin [H2(OBTPP)]. The demetallation was brought about by addition of perchloric acid(70%) 10ml to Cu(OBTPP) (250mg) in chloroform(150ml). The perchloric acid solution 70% was added to maintain a suitable pH for demetallation. The yellowish solution, obtained was stirred for about eight hours and the organic layer was separated and neutralised with 20% aqueous solution of sodium carbonate. The dark green coloured layer so obtained was separated and dried over anhydrous sodium sulphate. The solvent was removed leaving behind a dark green solid. This solid was dissolved in minimum quantity of chloroform and chromatographed on basic alumina column to obtain octabromotetraphenyl porphyrin[H2(OBTPP)](4).
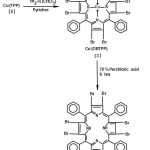 |
Figure 2 Click here to View figure |
Chlorooctabromotetraphenyl porphinato chromium [Cr(OBTPP)Cl]
The procedure used for the synthesis of Cr(OBTPP)Cl was as reported by Alder and co-werkers18. A typical synthesis is given below-
H2(OBTPP) (1.0gm) in 100ml DMF was taken in R.B.flask, and put to reflux so as to dissolve the ligand and after this CrCl3(0.2gm) was added to the refluxing solution. After refluxing the contents for 10minutes a further addition of CrCl3(0.2gm) was again added. The refluxing was continued for another 10minutes and an aliquot was taken from the reaction mixture and visible spectrum was recorded using CHCl3 as solvent. The spectrum indicated the presence of free base perphyrin H2(OBTPP) hence CrCl3(0.2gm) was again added to the solution and reflux continued for 10min. more. At this point a visible spectrum of an aliquot was taken from the reaction mixture which revealed the absence of any remaining free-base porphyrin. The refluxing was stopped and the reaction mixture was allowed to cool to room temperature. The cooled reaction mixture was poured into a flask-containing ice-cold water (500ml). The resulting precipitate was filtered and washed with small amounts of water and dried in oven for 1hr. at 1000C, the yield of crude [Cr(OBTPP)Cl] was 1.2gm.
Purification of Cr(OBTPP)Cl
Crude Cr(OBTPP)Cl (1.2gm) was dissolved in CHCl3(150 ml) and was loaded over alumina column(80-200 mesh). The column was eluted using CHCl3 as eluent. The unreacted H2(OBTPP) was eluted as green-red band with the solvent front and was followed by a slow-moving green band. As the green band came off the column it was monitored by visible spectroscopy. The band prior to Cr(OBTPP)Cl was eluted from the column was disearded. The volume of solvent was reduced to about 150ml using a rotary evaportor. Aqueous HCl(5ml) was added to the chloroform solution and stirring continued overnight. The solvent was removed on a rotary evaporator. The solid was recrystallized from chloroform / hexane and dried overnight in an oven. The yield of purified Cr(OBTPP)Cl after chromatography was 0.75gm.
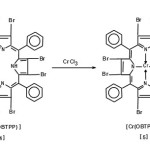 |
Figure 3 Click here to View figure |
Results and Discussion
Elemantal Analysis
The porphyrin complexes of chromium(III) derivatives so synthesized were subjected to chemical analysis. The results fall within the range expected for the proposed structures for these complexes(Table – I). It is seen that the observed values of the percentage of carbon, hydrogen and nitrogen of these complexes are in close agreement with the values calculated for these complexes.
Electronic spectra
The complex and porphyrin free bases were characterised using UV-visible spectroscopy. The electronic spectra of some complexes and free bases in the range 300-700nm were recorded and the values are given in (Table-II).
The products [TPP and Cu(TPP)] are purple and dark red crystals, respectively19. Porphyrin macrocycles exhibit two main bands in UV-visible spectra, the soret (a more intense band around 400 nm) and the Q-bands 500-650nm.
The appearance of Cu(TPP) and TPP differed in that Cu(TPP) was more of a dark red color and TPP was more of a vibrant purple color. The reason for this is that the intensity for TPP is much higher in the 600-700nm range (~200 a.u. at 654nm) compared to the 60a.u. at 654nm of Cu(TPP). A higher intensity in the red range results in a compound more resembling blue, which is why Cu(TPP) is redder than TPP. Metallated porphyrins such as Cu(TPP) are more symmetrical, resulting in fewer Q- bands and a simpler spectrum. The Soret Band is the highest peak in the UV-visible spectrum, around 400nm, and the Q-bands are the smaller bands in the spectrum around 500-650nm.
UV-visible spectra of Copper(II) in H2(OBTPP) which leads to formation of Cu(OBTPP) shows absorption bands at 426, 553, 586nm having a λmax value at 485nm. Cu(OCTPP) shows absorption at 448nm and 580nm. The shift is in agreement with the presence of electronegative halogen atom on these complexes as halogen are known to shift the absorption bands to longer wavelength. Cu(OCOBTPP) shows sharp peaks at 568nm, 615nm and also around 660nm with a shoulder band at 476nm. The wide variation of Cu(OCOBTPP) and its free base suggest that the complex has been formed.
The H2(OBTPP) displays absorption bands at 372 and 466nm with well formed weak bands at 568 and 627nm. H2(OCTPP) shows band at 372, 408, 514, 586nm. The lmax value was observed at 469nm in the case of H2(OCOBTPP) the band were obtained at 464, 556, 598 and 644nm respectively, with lmax value at 464nm. The comparison of the spectra of the free bases show that substitution of Br atom at β-pyrrole position leads to greater shift than substitution of electronegative group in the phenyl ring.
The introduction of Cr(III) in H2(OBTPP) which leads to formation of Cr(OBTPP)Cl shows absorption bands at 384,497, 603 nm having a lmax value at 497nm. Cr(OCTPP)Cl shows absorption at 386, 449, 565 and 643nm with a lmax value at 449 nm.
Magnetic Moment
Chromium(III) complexes show values in the range 3.79 B.M. indicating an octahedral structure (Table-I).
(Table1: Chemical Free Bases And Their Chromium(Iii) Complexes, Analysis Datas Of Porphyrin
|
S.NO.
|
PORPHYRIN |
ANALYSIS DATA |
MAGNETIC MOMENT μeff / B. M. |
||
|
C% |
H% |
N% |
|||
|
1. |
H2(OBTPP) |
42.20(42.29) |
1.80(1.77) |
4.28(4.49) |
– |
|
2. |
H2(OCTPP) |
59.15(59.36) |
2.32(2.49) |
6.24(6.28) |
– |
|
3. |
H2(OCOBTPP) |
34.32(34.71) |
0.78(0.92) |
3.45(3.68) |
– |
|
4. |
Cr(OBTPP)Cl |
39.40(39.65) |
1.25(1.65) |
4.60(4.32) |
3.79 |
* Calculated values are given in parentheses
(Table2: Electronic Spectral Datas Of Some Free Base Porphyrins And Metal Derivatives
|
S.NO. |
PORPHYRIN |
ABSORPTION PEAKS ( nm) |
|
1. |
H2(OBTPP) |
372, 466 (s), 568, 627 |
|
2. |
H2(OCTPP) |
372 (sh), 408 (s), 514, 586 |
|
3. |
H2(OCOBTPP) |
464 (s), 556, 598 (sh), 644 |
|
4. |
H2(TPP) |
420(s), 510, 545 |
|
5. |
Cu(TPP) |
417(s), 541, 580 |
|
6. |
Cu(OBTPP) |
426 (s), 553, 586 |
|
7. |
Cu(OCTPP) |
448 (s) , 580 |
|
8. |
Cu(OCOBTPP) |
476(sh), 568(s), 615,660 |
|
9. |
Cr(OBTPP)Cl |
384, 497 (s), 603 |
S=Strong, Sh=Shoulder
References
- Inamo, M.; Nakajima,K.; Bull.Chem.Soc.Japan. 1998, 71, 883
- Yaul,S.R.; Yaul,A.R.; Pethe,G.B.; Aswar,A.S.; Am.Eur.J.Sci.Res. 2009, 4,4,229.
- Abdou,S.; Tabi,T.; Raffat I.M.;Morsi,M.A.; Trans.Met.Chem. 2004, 29, 543
- Prasad, R.N.; Mathur,M.; J.Serb.Chem.Soc. 2002, 67, 12, 825
- Ellis,P.E.; Lyons,J.E.; J.Chem.Soc.Chem.Commun.1989, 1315
- Dolphin,D.; Felton,R.H.; Acc.Chem.Res.1974,7,26
- Liao,M.S.; Scheiner,S.; J.Chem.Phy.2002,117,1,205
- Holm,R.S.; Chem.Rev.1987.87.1401
- Ellis,P.E.; Lyans,I.E.; U.S.Patent.1997.667.328
- Lane,B.S.; Burgess,K.; Chem.Rev.2003.103.2457
- Reddy,C.; Hamilton,G.A.; Madystha,K.M.; Eds.Academic Press.New York.1990
- Sheldon,R.A.; Kochi,J.K.; Metal catalyzed oxidation of organic compound. Academic press. New York.1981
- Mijs,W.J.; De jonge,C.R.I.; Plenum Press. New York.1986
- Spadavecchia,J.; Ciccarella,G.; Siciliano,P.; Capone,S.; Rella,R.; J.Mol. Catel.A. 2004, 100,88
- Dorough,G.D.; MillerJ.R.; Huennekens,F.M.; J.Am.Chem.Soc. 1951,73,4315
- Alder,A.D.; Longo,F.R.; Vardi,V.; Inorg.Synth. 1976, 16, 213
- Bhyrappa,P.; Krishnan,V.; J.Chem.Sci. 2004, 116, 2, 71
- Bhyrappa,P.; Krishnan,V.; Inorg.Chem. 1991, 30, 239
- Biesaga,M.; Pyrzynska,K.; Trojanowicz,M.; Talanta. 2000, 51, 209

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


