A comparative of quantum mechanical Calculations on adsorption of CCl2O by Carbon-Carbon & Aluminum -Nitride Nanotubes
A. Kazemi Babaheydari*1
Department of chemistry, Islamic Azad University, Shahrekord Branch, Shahrekord, Iran Corresponding Author kazemiali@iaushk.ac.ir
DOI : http://dx.doi.org/10.13005/ojc/310209
Article Received on :
Article Accepted on :
Article Published : 24 Jun 2015
In this paper, is calculated structural optimization and interactions on surface nanotubes (AlN,CNT) and phosgene. Based on the optimized ground state geometries using B3LYP/6-31G* method, the NBO analysis of donor-acceptor (bond- anti bond) interactions revealed that the stabilizationenergies associated with the electronic delocalization.
KEYWORDS:molecular orbital (MO); B3LYP; Natural Bond Orbital (NBO); phosgene; Aluminum -Nitride
Download this article as:| Copy the following to cite this article: Babaheydari A. K. A comparative of quantum mechanical Calculations on adsorption of CCl2O by Carbon-Carbon & Aluminum -Nitride Nanotubes. Orient J Chem 2015;31(2). |
| Copy the following to cite this URL: Babaheydari A. K. A comparative of quantum mechanical Calculations on adsorption of CCl2O by Carbon-Carbon & Aluminum -Nitride Nanotubes. Available from: http://www.orientjchem.org/?p=9410 |
Introduction
Discovery of carbon nanotubes [1] has sparked intense research activity within the past decade. These novel materials have a wide range of potential applications ranging from the field of nano electronics to nano scale bio technology. They may be used as molecular field-effect transistors [2,3] electron Field emitters [2,4], artificial muscles [2,5] , or even DNA sequencing agents [6]. The adsorption behavior of single atoms or gas molecules on carbon nanotubes has been studied extensively in the past decade by experiments [7-9] and theoretical calculation [10-14]. Aluminum nitride nanotubes (AlNNTs) are inorganic analog carbon nanotubes (CNTs). They are isoelectronic with CNTs and have been synthesized successfully by different research groups [5–7]. Because of their high temperature stability, large energy gap, thermal conductivity and low thermal expansion [8], AlNNTs and aluminum nitride nano materials are widely used in technological applications, mainly in micro and optoelectronics such as laser diodes and solar-blindltraviolet photo detectors and semiconductors [8]. Unlike CNTs, AlNNTs exhibit electronic properties and semiconductor behavior independent of length, tubular diameter and chirality. Tuning the electronic structures of the semiconducting AlNNTs for specific applications important in building specific electronic and mechanical devices. Phosgene is a major component of natural gas and its adsorption behavior in pores has been extensively studied. In this study, B3LYP studies of the absorption behavior of phosgene gas on nanotube were performed in terms of adsorption energy.
Computational details
In our current study, extensive quantum mechanical calculations of structure of Aluminum -Nitride nanotube [zigzag (8,0)] have been performed on a Pentium-4 based system using Gaussian 03 program [17] . At first, we have modeled and the nanotube with Nanotube Modeler package and then optimized at the B3LYP level of theory with 6-31G*basis set. After fully optimization of nanotube, we have calculated adsorption of phosgene at the level of 6-31G* theory on the outside (external) of carbon nanotube and have been reported in Table 1, and finally we calculate Natural Bond Orbital (NBO) parameters for this structure, Table 2. The BE (Binding Energy) of CCl2O on the optimized nanotubes model is calculated as follows:
BE=ECCl2O-AlNNT – [EAlNNTs+ECCl2O] Eq(1)
BE=ECCl2O–CNNT – [E CNNT +ECCl2O] Eq(2)
Where ECCl2O–AlNNTs was obtained from optimization of the adsorption CCl2O on surfaces AlN models, AlNNTs is the energy of the optimized AlNNTs structure and CNTs energy of the optimized CNT s structure and ECCl2O the energy[13] of the optimized phosgene gas.
Results and Discussion
In this paper B3LYP method with 6-31G*basis set were employed to investigate the structure optimization, energy minimization of AIN, carbon nanotubes and phosgene (Fig 1 and Table 1).
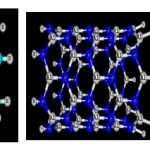 |
Figure1: The final optimized of AlN and Carbon nanotubes [zigzag (8,0 ) ] with 12nm length and phosgene gas obtained through B3LYP (6-31G*) calculation Click here to View figure |
Table1: Calculated energy values (hartree) of AlN (Aluminum –Nitride), Carbon-carbon nanotubes and phosgene in gas phase at the level of B3LYP /6-31G*
|
parameters |
Phosgene |
AlN |
CNT |
|
E(total) |
-1033/714 |
-11901.3761 |
-3058.01718 |
|
EHOMO/ev |
-0/360 |
-0.2348 |
-0.1348 |
|
ELUMO/ev |
0/310 |
-0.0706 |
-0.1243 |
|
[I=-EHOMO]/ev |
0/360 |
0.2348 |
0.1348 |
|
[A=-LUMO]/ev |
-0/310 |
0.0706 |
0.1243 |
|
[ŋ=(I-A)/2]/ev |
0/335 |
0.0821 |
0.0052 |
|
[µ=-(I+A)/2]/ev |
-0/025 |
-0.1527 |
-0.1295 |
|
[s=1/2ŋ]/ev-1 |
0/167 |
0.0410 |
0.0026 |
|
[w=µ2/2ŋ]ev |
0/0001 |
0.5687 |
0.000 |
after fully optimization of AlN and Carbon nanotubes [zigzag (8,0 )], we had calculated optimized structure of interaction between nanotube and phosgene at the level of B3LYP /6-31G*theory (Fig 2),: and then performed Natural Bond Orbital(NBO) calculations for give NBO important parameters, Table2
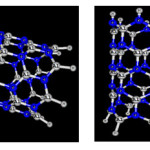 |
Figure2: Adsorption phosgene by AlN nanotube in two position (0-down, Cl-down), optimized by B3LYP /6-31G*basis set Click here to View figure |
To study absorption behavior of phosgene gas on AlN nanotube, we performed absorption for two sites in nanotube (outside (external) of carbon nanotube) the after a full optimization (Table 1), we found two structures of this absorption (Fig 2, 3)
Table1: Calculated energy values (hartree) of AlN (Aluminum –Nitride), Carbon-carbon nanotubes and phosgene in gas phase at the level of B3LYP /6-31G*
I=ionization potential, A=electron affinity, ŋ=Global hardness, μ=chemical potential and w= electrophilicity
In the NBO analysis, in order to compute the span of the valence space, each valence bonding NBO (σAB), must in turn, be paired with a corresponding valence anticoding NBO (σ*AB) Namely, the Lewis σ-type (donor) NBO are complemented by the non-Lewis σ*-type (acceptor) NBO that are formally empty in an idealized Lewis structure picture. Readily, the general transformation to NBO leads to orbitals that are unoccupied in the formal Lewis structure. As a result, the filled NBO of the natural Lewis structure are well adopted to describe covalence effects in molecules. Since the non-covalent delocalization effects are associated with σ → σ*interactions between filled (donor) and unfilled (acceptor) orbitals, it is natural to describe them as being of donor–acceptor, charge transfer, or generalized “Lewis base-Lewis acid” type.
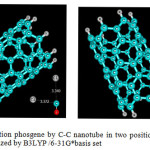 |
Figure3: Adsorption phosgene by C-C nanotube in two position (0-down, Cl-down), optimized by B3LYP /6-31G*basis set Click here to View figure |
The anti-bonds represent unused valence-shell capacity and spanning portions of the atomic valence space that are formally unsaturated by covalent bond formation. Weak occupancies of the valence ant bonds signal irreducible departures from an idealized localized Lewis picture, i.e. true “delocalization effects”.As a result, in the NBO analysis, the donor–acceptor (bond–anti bond) interactions are taken into consideration by examining all possible interactions between ‘filled’ (donor) Lewis-type NBO and ‘empty’ (acceptor) non-Lewis NBO and then estimating their energies by second-order perturbation theory.
Table2: Calculated energy values (hartree) adsorption phosgene gas with C-C (Carbon –Carbon ) & Al-N (Aluminum –Nitride ) nanotubes in gas phase at the level of B3LYP /6-31G*
|
parameters |
Phosgene-AlNNT |
Phosgene-CNT |
||
|
Cl-down |
O-down |
Cl-down |
O-down |
|
|
Eads |
2975.4562 |
-0.0097 |
-0.01432 |
0.00138 |
|
EHOMO/ev |
-0.2365 |
-0.2288 |
-0.1598 |
-0.1309 |
|
ELUMO/ev |
-0.0787 |
-0.1104 |
-0.1394 |
-0.12027 |
|
[I=-EHOMO]/ev |
0.2365 |
0.2288 |
0.1598 |
0.1309 |
|
[A=-LUMO]/ev |
0.0787 |
0.1104 |
0.1394 |
0.12027 |
|
[ŋ=(I-A)/2]/ev |
0.0789 |
0.0592 |
0.0102 |
0.0053 |
|
[µ=-(I+A)/2]/ev |
-0.1576 |
-0.1696 |
-0.1496 |
-0.2512 |
|
[s=1/2ŋ]/ev-1 |
0.03945 |
0.0296 |
0.0051 |
0.0026 |
|
[w=µ2/2ŋ]ev |
0.00097 |
0.0008 |
0.0001 |
0.00016 |
I=ionization potential, A=electron affinity, ŋ=Global hardness, μ=chemical potential and w=electrophilicity
These interactions (or energetics stabilizations) are referred to as ‘delocalization’ corrections to the zeroth-order natural Lewis structure. The most important interaction between “filled” (donor) Lewis-type NBO and “empty” (acceptor) non-Lewis isreported in Table (2,3) in Fig 3. We observed interactions between the bonding orbital (σ) C20-C50 and H49-C29 but do not this bonding orbital for phosgene.
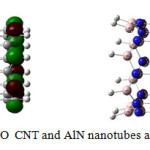 |
Figure3: HOMO-LUMO CNT and AlN nanotubes after optimization Click here to View figure |
Table3: The second-order perturbation energies E(2) (kcal/mol) corresponding to the most important charge transfer interactions (donor → acceptor) in the compounds studied by using B3LYP /6-31G*method for Al29-N49
| Complex(Al-N, phosgene) | Donor → Acceptor |
E(2), kcal/mol |
– |
F(i,j) |
||
|
O-down |
LP(Cl100) | (N3-Al73) |
0.11 |
0.67 |
0.008 |
|
|
σ*(N3-Al77) |
0.99 |
1.07 |
0.039 |
|||
|
σ*(N3-Al92) |
0.19 |
1.25 |
0.014 |
|||
| (C97-O98) | σ*(N37-Al92) |
0.16 |
0.37 |
0.016 |
||
|
σ*(N2-Al92) |
0.29 |
0.37 |
0.022 |
|||
| σ(N2-Al81) | σ*(C97-O98) |
0.28 |
0.12 |
0.005 |
||
| σ(C97-O99) | σ*(C97-O98) |
4.38 |
0.38 |
0.037 |
||
|
Cl-down |
LP(Cl100) |
σ*(N3-Al73) |
0.12 |
0.70 |
0.01 |
|
|
σ*(N3-Al77) |
0.98 |
1.09 |
0.049 |
|||
|
σ*(N3-Al92) |
0.21 |
1.20 |
0.017 |
|||
| (C97-O98) |
σ*(N37-Al92) |
0.18 |
0.39 |
0.014 |
||
|
σ*(N2-Al92) |
0.32 |
0.39 |
0.023 |
|||
| σ(N2-Al81) |
σ*(C97-O98) |
0.29 |
0.15 |
0.006 |
||
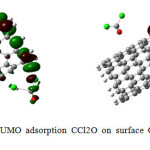 |
Figure4: HOMO-LUMO adsorption CCl2O on surface CNT nanotube after optimization Click here to View figure |
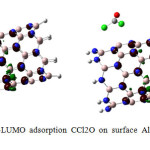 |
Figure4: HOMO-LUMO adsorption CCl2O on surface AlN nanotube after optimization Click here to View figure |
Table4: The second-order perturbation energies E (2) (kcal/mol) corresponding to the most important charge transfer interactions (donor → acceptor) in the compounds studied by using B3LYP /6-31G*method for C61H12Cl2O
| Complex(C-C, phosgene) | Donor → Acceptor |
E(2), kcal/mol |
– |
F(i,j) |
||
|
O-down |
(C97-CL99) | (C97-Cl99) |
0.56 |
0.82 |
0.020 |
|
| σ*( C97-Cl100) |
1.55 |
0.82 |
0.033 |
|||
|
σ*( C97-O98) |
0.59 |
0.4 |
0.015 |
|||
| LP(O98) | σ*(C97-Cl99) |
40.60 |
0.38 |
0.114 |
||
|
σ*( C97-Cl99) |
2.48 |
0.80 |
0.041 |
|||
| LP(Cl99) | σ*(C97-O98) |
1.90 |
1.52 |
0.048 |
||
| σ*(C97-O98) | σ*(C6-C75) |
0.22 |
0.10 |
0.007 |
||
|
Cl-down |
(C97-CL99) | (C97-Cl99) |
0.58 |
0.75 |
0.025 |
|
| σ*( C97-Cl100) |
1.57 |
0.82 |
0.038 |
|||
|
σ*( C97-O98) |
0.6 |
0.4 |
0.019 |
|||
| LP(O98) | σ*(C97-Cl99) |
40.61 |
0.38 |
0.116 |
||
|
σ*( C97-Cl99) |
2.45 |
0.80 |
0.043 |
|||
| LP(Cl99) | σ*(C97-O98) |
2.1 |
1.52 |
0.049 |
||
| σ*(C97-O98) | σ*(C6-C75) |
0.27 |
0.11 |
0.006 |
||
Conclusion
we have modeled the nanotube with Nanotube Modeler program and then optimized at the B3LYP level of theorywith 6-31G*basis set. After fully optimization of nanotube, we have calculated adsorption of phosgene at the levelof 6-31G*theory on outside (external) of Aluminum -Nitride nanotube.
The most important interaction between “filled” (donor) Lewis-type NBO and “empty” (acceptor) non-Lewis, weobserved between the bonding orbital (σ) C20-C50 and H49-C29 but don’t see this bonding orbital for nano tube and internal phosgene.
Acknowledgment
This work was supported by Islamic Azad University Shahrekord Branch, we thank him.
Reference
- S.Iijima , Nature 1991, 354, 56.
- A.Mine_, K.Atkinson, S.Roth, Carbon Nanotube, in:F.Sch¨uth, S.W.Sing, J. Weitkamp (Eds.), Handbook of Porous Solids, Wiley-VCH, Weinheim, 2002.
- Z.Yao, H.W.C.Postma, L.Balents, C.Dekker, Nature 1999, 402, 273.
- O.Zhou, H. Shimoda, B. Gao, S. Oh, L. Fleming, G. Yue, Acc. Chem. Res. 2002, 35, 1045.
- R.H.Baughman, C.Cui, A.A.Zakhidov. Z.Iqbal, J.N.Barisci, G.M.Spinks, G.G.Wallace, A.Mazzoldi, D.DeRossi, A.G.Rinzler, O. Jaschinski, S.Roth, M.Kertesz, Science 1999, 284, 1340.
- H.Gao, Y.Kong, D.Cui, NanoLe 2003, 3, 471.
- J. Kong, N.R. Franklin, C.W. Zhou, M.G. Chapline, S. Peng, K.J. Cho, H.J. Dai, Science 2000, 287, 622.
- F. Villalpando-Paez, A.H. Romero, E. Munoz-Sandoval, L.M. Martinez, H. Terrones, M. Terrones, Chem. Phys.Le_.386 (2004) 137.
- L. ValenEni et al., Chem. Phys. Le_. 2004, 387, 356.
- S. Peng, K.J. Cho, Nanotechnology 11 (2000) 57.
- X. Lu, Z.F. Chen, P.V. Schleyer, J. Am. Chem. Soc. 2005, 127, 20.
- L.V. Liu, W.Q. Tian, Y.A. Wang, J. Phys. Chem. B 2006, 110, 13037.
- F. Ding, K. Bolton, A. Rosen, J. Phys. Chem. B 2004, 108, 17369.
- J. Zhao, A. MarEnez-Limia, P.B. Balbuena, Nanotechnology 2005, 16, S575.
- L.V. Liu, W.Q. Tian, Y.A. Wang, J. Phys. Chem. B 2006, 110, 13037.
- ShiminHou ,Ziyong Shen , Xingyu Zhao , Zengquan Xue. Chemical Physics Leters 2003, 373, 308-313.
- Gaussian 98, Revision A.7,Frisch.M.J; Trucks. G..W;Schlegel.H.B;Scuseria. G.E;Robb.M.A; Cheeseman. J. R; ZZakrzewski. V.G; ontgomery. J. A; Stratmann;R.E; BurantJ. C; Apprich. S; Millam. J.M; Daniels. A.D; Kudin. K. N; Strain. M. C; Farkas. O; Tomasi. J; Barone. V; Cossi. M; Cammi. R; Mennucci. B; Pomelli. C; Adamoc; Clifford. S; Ochterski. J; Petersson. G.A;Ayala.P.Y; Cui.Q; Morokuma.K;Malick. D.K;Rabuck.A.D;Raghavachari.K; Foresman. J.B;Cioslowski. J; Ortiz.J.V; Baboul. A.G; Stefanov. B.B; LIU.G;Liashenko. A; Piskorz. P; Komaromi. I; Gomperts.R;Martin.R.L;A;Fox. D. J; Keith. T; Al-laham.M.Peng. C. Y ;Nanayakkara .A;Gonzalez.C; Challacombe. M; Gill.P.M.W;Johnson. B; Chen.W; Wong.M.W;Andres.j. L; Gonzalez.C; Head-Gordon. M; Replogle.E.S and Pople.J.A, Gaussian, Inc; Pi_sburgh PA, 1998.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


